Autosomal dominant frontometaphyseal dysplasia: Delineation of the clinical phenotype
Abstract
Frontometaphyseal dysplasia (FMD) is caused by gain-of-function mutations in the X-linked gene FLNA in approximately 50% of patients. Recently we characterized an autosomal dominant form of FMD (AD-FMD) caused by mutations in MAP3K7, which accounts for the condition in the majority of patients who lack a FLNA mutation. We previously also described a patient with a de novo variant in TAB2, which we hypothesized was causative of another form of AD-FMD. In this study, a cohort of 20 individuals with AD-FMD is clinically evaluated. This cohort consists of 15 individuals with the recently described, recurrent mutation (c.1454C>T) in MAP3K7, as well as three individuals with missense mutations that result in substitutions in the N-terminal kinase domain of TGFβ-activated kinase 1 (TAK1), encoded by MAP3K7. Additionally, two individuals have missense variants in the gene TAB2, which encodes a protein with a close functional relationship to TAK1, TAK1-associated binding protein 2 (TAB2). Although the X-linked and autosomal dominant forms of FMD are very similar, there are distinctions to be made between the two conditions. Individuals with AD-FMD have characteristic facial features, and are more likely to be deaf, have scoliosis and cervical fusions, and have a cleft palate. Furthermore, there are features only found in AD-FMD in our review of the literature including valgus deformity of the feet and predisposition to keloid scarring. Finally, intellectual disability is present in a small number of subjects with AD-FMD but has not been described in association with X-linked FMD.
1 INTRODUCTION
Frontometaphyseal dysplasia (FMD) is a sclerosing skeletal dysplasia clinically characterized by prominent supraorbital ridges, sclerosis of the skull, and dense, undermodelled cortices of the long bones and phalanges. A range of extraskeletal manifestations, such as progressive joint contractures, genitourinary tract defects, laryngeal stenosis, and hearing loss, significantly impact the health and wellbeing of affected individuals over their lifetime (Fitzsimmons, Fitzsimmons, Barrow, & Gilbert, 1982; Morava, Illes, Weisenbach, Karteszi, & Kosztolanyi, 2003; Robertson et al., 2006). Furthermore, some individuals have a predisposition to develop keloid scars which can be disfiguring and debilitating (Basart et al., 2015; Wade et al., 2016). To date, little data is available on the phenotype of individuals with FMD, especially those living into their fourth decade and beyond.
FMD exhibits locus heterogeneity. A proportion of individuals have gain-of-function mutations in FLNA, which encodes the actin binding protein, filamin A (Robertson et al., 2003). Due to the X-chromosomal location of FLNA, the presentation of FMD is more severe in males and milder in females. Nevertheless there is considerable variation in the presentation in both sexes, which in the instances of females may be related to the degree of skewing of X-inactivation (Robertson, 2007; Robertson et al., 2006). X-linked (XL)-FMD is allelic to a spectrum of phenotypically related skeletal dysplasias, the otopalatodigital spectrum disorders (OPDSDs), which are also caused by gain-of-function mutations in FLNA (Robertson et al., 2003). Mutations in FLNA which lead to FMD and the OPDSDs tend to be missense or, small, in-frame deletions which are found in specific domains of the protein (Robertson, 2007; Robertson et al., 2003).
Approximately 50% of FMD patients do not have their condition explained by mutations in FLNA (Basart et al., 2015; Robertson et al., 2006). We have recently characterized mutations in MAP3K7, encoding TGFβ-activated kinase 1 (TAK1), that are causative of an autosomal dominant type of FMD. Consequently we suggested that FMD caused by mutations in FLNA be referred to as FMD1 and, that caused by mutations in MAP3K7 be referred to as FMD2 (Wade et al., 2016). We also described a single individual with a de novo mutation in TAB2, encoding TAK1-associated binding protein 2 (TAB2), which we predicted would most likely represent a third locus for the disorder (Wade et al., 2016). We hypothesized that this missense mutation would be causative of the disease because of the close functional relationship between TAK1 and TAB2 (Besse et al., 2007; Wade et al., 2016). Evidence from knockout mouse models (Greenblatt et al., 2013; Qi et al., 2014; Sanjo et al., 2003) as well as from in vitro work (Wade et al., 2016) suggests that mutations in MAP3K7 and TAB2 also produce the FMD phenotype through a gain-of-function mechanism.
The most common form of autosomal dominant (AD)-FMD is caused by a recurrent MAP3K7 mutation, c.1454C>T (GenBank: NM_003188.3), that predicts a substitution in TAK1, p.Pro485Leu (Wade et al., 2016). This mutation results in a FMD phenotype which is remarkably similar to that exhibited by the single patient with the TAB2 de novo variant. As well as the recurrent C-terminal substitution, variants in the N-terminal kinase domain of TAK1 also confer an FMD phenotype, albeit with a milder presentation.
Here we describe the clinical presentation of AD-FMD attributed to mutations in MAP3K7 and TAB2. The presentation of AD-FMD is distinct from FMD caused by mutations in FLNA, however the phenotypes are similar enough to suggest that the mechanism of disease is related to the same biochemical process.
2 MATERIALS AND METHODS
2.1 Patient ascertainment
Patients were recruited based on clinician-initiated referral and consented under approved protocols MEC/08/08/094 and 13/STH/56 (Health and Disability Ethics Committee, New Zealand). The diagnosis of FMD was confirmed based on published clinical and radiological criteria (Robertson et al., 2006). FLNA mutations were sought using Sanger sequencing to examine the exons and exon-intron boundaries of that gene. Copy number variants of FLNA were excluded by use of a customized multiplex ligation-dependent probe amplification (MLPA) probeset. Cases with a firm clinical diagnosis of FMD and no detectable mutation in the exons or intron exon boundaries of FLNA formed a cohort of 20 individuals. Thirteen individuals have been clinically reported before (Table 1) (Basart et al., 2015; Morava et al., 2003; Robertson et al., 2006). Nineteen individuals (all except individual 09) were described as part of a biochemical study of AD-FMD (Wade et al., 2016). We are including all known instances of AD-FMD here for a clarity.

- U, unknown; na, not applicable; D, deceased; presented previously in *(Robertson et al., 2006).
- All individuals, except for 09, were described in (Wade et al., 2016). Where possible, individuals have been clinically re-evaluated therefore this table is this most up-to-date phenotypic spectrum.
- Protein substitution. see Supplementary Table SI for full mutation information.
- + Described in Present.
- − Described inAbsent.
- †(Basart et al., 2015).
- ‡(Morava et al., 2003).
- 1Cervical spinal cord compression.
- 2Aortic dilatation.
2.2 Mutation discovery
Previously we utilized whole exome sequencing and targeted Sanger sequencing (Wade et al., 2016) to undertake mutation discovery on 19 participants. Subsequently a sample from individual 09 (Table 1) was subjected to exome sequencing using the Illumina HiSeq (BGI-Europe, Denmark) platform after enrichment of the exome with the Agilent SureSelectXT Human All Exon 50 Mb capture kit. After read alignment with BWA and variant calling with GATK, variants were annotated by the Genetics Department of the Radboud UMC using an in-house analytical pipeline. Candidate variants were confirmed with Sanger sequencing.
3 RESULTS
3.1 Craniofacial findings
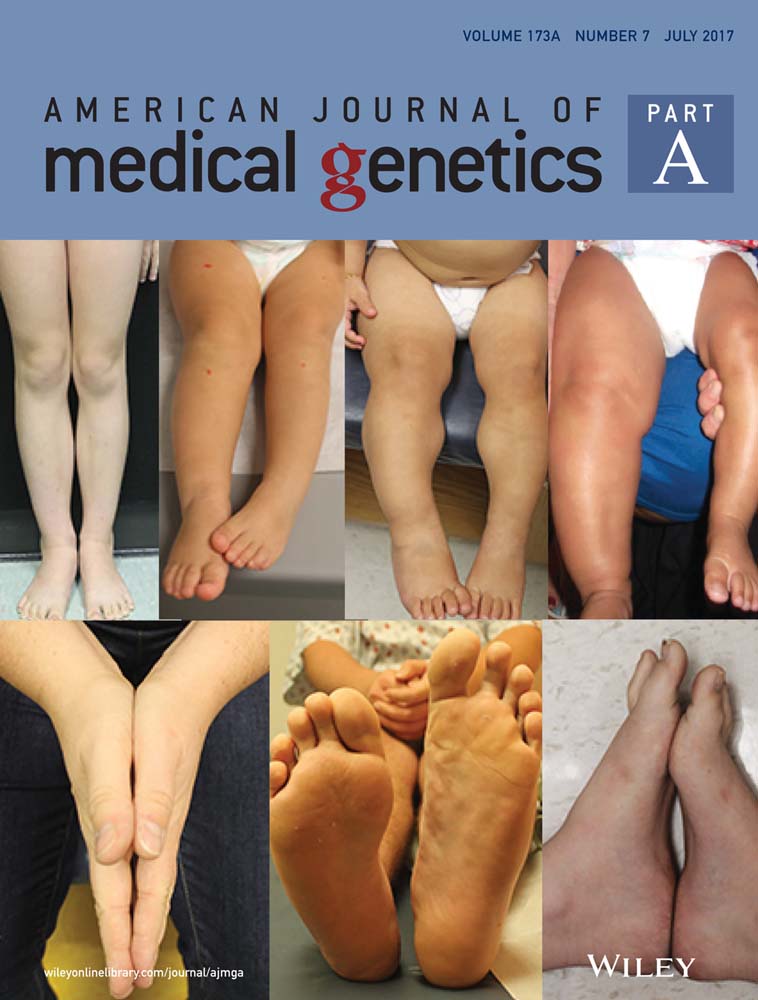
The 20 subjects with FMD described here all manifested the three mandatory diagnostic facial features of FMD; prominent supraorbital ridges, downslanting palpebral fissures, and hypertelorism (Robertson et al., 2006) (Figure 1, Table 1). A broad nasal bridge was a ubiquitous finding (20/20). Despite manifesting similar facial characteristics as seen in XL-FMD, patients with the recurrent C-terminal substitution in TAK1 (p.Pro485Leu), have coarser facial features than those individuals with XL-FMD (Figure 1). However those individuals with mutations in the 5′ region of MAP3K7, which encodes the kinase domain of TAK1 (patients 07, 19, and 20; Table 1 and Supplementary Table S1) manifest notably milder facial features (Figure 1, Table 1). Micrognathia, a mandatory diagnostic feature of XL-FMD, was only present in 16/20 (80%) of the individuals with TAB2 and MAP3K7 mutations (Table 1). Other features which may set AD-FMD apart from XL-FMD include the lack of bias toward affected males and the presence of hearing loss, which can be conductive, sensorineural or mixed. Some kind of deafness is observed in 6/8 (75%) males and 11/11(100%) females in this cohort compared to 67% of males and 27% of females with XL-FMD (Robertson et al., 2006) (Table 1). Cleft palate or a bifid uvula is more common in AD-FMD, occurring in 6/19 (32%) patients presented here compared to 5% of XL-FMD patients (Robertson et al., 2006) (Table 1). Radiological findings in the skull in those with AD-FMD were indistinguishable from those seen in XL-FMD for the majority of individuals, the exception being two individuals (07 and 19) with the milder presentation of AD-FMD due to substitutions in the kinase domain of TAK1: p.Gly168Arg (MAP3K7 c.502G>C) and p.Glu70Gln (MAP3K7 c.208G>C), who lacked skull-base sclerosis (Table 1 and Supplementary Table S1).

3.2 Limb and digit manifestations
Contractures of the fingers, wrists, and elbows are an important diagnostic feature of both XL-FMD and AD-FMD. In this AD-FMD cohort, 19/20 had digital (Figure 2a,c) and wrist contractures and 19/19 had either a contracture of the elbow joint or a dislocated radial head (Table 1). Ulna deviation of the hands, which is a mandatory diagnostic criterion of XL-FMD, was seen in 13/18 in this cohort (Table 1). There are other notable differences in the hands and feet of AD-FMD patients; the fingers and thumbs are long (as they also are in XL-FMD; Figure 2a,b, Table 1) but the spatulate finger and thumb tips seen in XL-FMD are not as prevalent. Additionally a valgus deformity of the feet was seen in 8/15 subjects (Table 1), a sign not previously associated with XL-FMD. Radiologically, undermodeling of the phalanges is a distinct finding (Figures 2b,d). The long bones can be slightly bowed, with dense cortices. Furthermore the femur and tibia display abnormal modeling of the diaphysis and metaphysis giving the appearance of an Erlenmeyer-flask (Figure 2e, Table 1).

3.3 Other radiological characteristics
Other radiological features which may help distinguish XL-FMD from AD-FMD include scoliosis, present in 16/20 (80%) with AD-FMD (Figure 3a, Table 1) denoting it as more prevalent in AD-FMD than in the X-linked condition (56% of males and 18% of females (Robertson et al., 2006)), an observation not previously noted in studies of FLNA mutation-negative FMD (Morava et al., 2003; Robertson et al., 2006). Another radiological finding which may be more prevalent in AD-FMD is the presence of cervical vertebral fusions, seen in 12/19 (63%) individuals in this cohort (Figure 3b, Table 1). Cervical fusions are only seen in 33% of FLNA-mutation positive males, and have never been reported in FLNA mutation-positive females (Robertson et al., 2006). Notably one individual with a MAP3K7 mutation developed a cervical spine myelopathy secondary to cervical vertebral anomalies.

3.4 Non-skeletal findings
The extraskeletal manifestations of FMD result in significant morbidity and are often the cause of early mortality. Individuals with AD-FMD manifest many of the same extraskeletal malformations as the patients with XL-FMD without the bias toward an increased prevalence in males. Subglottic stenosis or congenital stridor was seen in 6/16 (38%) individuals studied here (Table 1) compared to 11% of males and 9% of females with XL-FMD (Robertson et al., 2006). Genitourinary tract defects, usually in the form of urethral obstruction, were seen in 3/8 (38%) of the male patients described here (Table 1) and have previously been reported in 22% of males with XL-FMD. There are no previous reports of such defects in females with XL-FMD (Robertson et al., 2006), however 2/11 (18%) female patients in this cohort have some form of genitourinary tract defect (Table 1). Structural heart defects which are sometimes present in XL-FMD males (22%) were found in 2/6 (33%) males and 4/10 (40%) females with AD-FMD. A small number of patients with dilation of the aorta, that was slowly progressive in one instance, were noted (Table 1). Finally, and significantly, intellectual disability was observed in 3/20 individuals with AD-FMD (Table 1) but has never been previously associated with FMD of any description (Robertson et al., 2006). Intellectual disability is only seen in the individuals with the TAK1 p.Pro485Leu substitution, and a further individual with intellectual disability has been reported with this substitution in another study (Martinez et al., 2016).
3.5 Predisposition to keloid scarring
Keloid scar formation is not a reported manifestation of XL-FMD but has been connected with another FLNA-associated phenotype, which manifests with cardiac and kidney malformations and joint contractures (Atwal et al., 2015; Lah et al., 2015). Individuals with the skeletal manifestations of FMD, together with an increased tendency to form keloid scars generally do not have a mutation in FLNA (Basart et al., 2015; Wade et al., 2016). In the AD-FMD cohort studied here 10/20 patients have some form of keloid scarring (Table 1) and this finding is more prevalent in females (7/10). The severity of the scarring varies considerably between individuals with one individual developing florid keloid after relatively trivial procedures such as venepuncture (Basart et al., 2015).
3.6 Genotype-phenotype correlation in AD-FMD
The most common FMD-causing mutation found in MAP3K7 Leads to the substitution p.Pro485Leu, and was observed in 15/20 (75%) individuals in this cohort. The other three mutations found in MAP3K7 predict substitutions in the N-terminal kinase domain of TAK1 (Supplementary Table S1) (Wade et al., 2016). In one case the mutation was de novo, in the other two unrelated cases the mutation was transmitted from the affected mother (Wade et al., 2016). Substitutions in the kinase domain lead to a notably milder presentation than the recurrent mutation (Figure 4, Table 1). The three individuals with kinase domain substitutions have less dramatic undermodeling of the phalanges, minimal splaying of the metaphyses and can lack skull base sclerosis (Figure 4, Table 1). None of these three individuals have developed keloid.

FMD has overlapping features with the recently described dominant condition cardiospondylocarpofacial syndrome (CSCF) which is associated with missense mutations, or small deletions, affecting the N-terminal region of TAK1 (Le Goff et al., 2016). These include facial characteristics such as hypertelorism and full cheeks, as well as deafness, cervical fusions, and cardiac defects (Le Goff et al., 2016); however clear clinical distinctions can be made between the two conditions. CSCF is characterized by brachydactyly, short extremities and joint laxity (Le Goff et al., 2016), in direct contrast to the FMD phenotype which, in contradistinction, exhibits arachnodactyly and joint contractures (Table 1). Furthermore the three patients with kinase domain substitutions have the characteristic prominent brow ridges which are absent in individuals with CSCF.
3.7 Individuals with mutations in TAB2
Here we describe two individuals with TAB2 substitutions p.Gln540Arg (TAB2 c.1619A>G; NM_ NM_015093.5) and p.Glu569Lys (TAB2 c.1705G>A), caused by variants in the same exon of TAB2. The p.Gln540Arg variant is newly described, not present in the Exome Aggregation Consortium (ExAC) database and the affected residue is phylogenetically conserved (Supplementary Figure S1). Variant effect prediction software predicted the p.Gln540Arg substitution as pathogenic: SIFT score of 0 (damaging), PolyPhen2 score of 0.954 (probably damaging), and predicted to be disease causing by MutationTaster. Both of the described variants in TAB2 had arisen de novo. The subject with this variant has a remarkably similar presentation to the original female we described with a TAB2 mutation (Figure 1), and both resemble the individuals with the recurrent MAP3K7 mutation more than those subjects with kinase domain substitutions in TAK1 (Figure 1). Neither subject with a TAB2 variant has keloid scars.
4 DISCUSSION
We have previously shown that FMD is a genetically heterogeneous disorder caused by mutations in FLNA (Robertson et al., 2003), MAP3K7 and TAB2 (Wade et al., 2016). Here we present the clinical phenotype of patients with mutations in these recently characterized AD-FMD loci. This cohort of patients with AD-FMD can be divided into three categories: those with the recurrent C-terminal substitution in TAK1 (p.Pro485Leu) which confers the most severe of the three phenotypes, those with substitutions in the TAK1 kinase domain which result in a milder presentation, and finally those patients with mutations that predict substitutions in the TAK1 interacting protein, TAB2. Previously we described a single individual with a typical AD-FMD phenotype and a de novo missense variant in TAB2 (Wade et al., 2016). We hypothesized that the resulting substitution (p.Glu569Lys) would eventually be shown to constitute the basis for her AD-FMD phenotype and that TAB2 would represent a third FMD locus. Here we present a second individual with a mutation in TAB2, in the same exon as the originally described variant. The presence of a novel missense mutation in TAB2, which confers a similar phenotype to that associated with the p.Glu569Lys substitution, is solid evidence that this gene represents a third FMD locus and a second cause of AD-FMD.
Despite considerable phenotypic overlap with XL-FMD which suggests that the two conditions are caused by disruption of the same biochemical pathway, distinctions can be made which may aid diagnosis. Clearly the inheritance pattern and presentation of females in the pedigree can indicate the likelihood or otherwise that X-linked inheritance of the trait might apply. Additionally, to our knowledge there are no instances of inheritance of the recurrent MAP3K7 mutation (c.1454C>T, TAK1 p.Pro485Leu), although two of the singular MAP3K7 mutations (c.208G>C, TAK1 p.Glu70Gln and c.299T>A, TAK1 p.Val100Glu) have been vertically transmitted from a clinically affected parent.
A common finding in all forms of FMD is undermodeling of the phalanges reflecting that an X-ray of the hands is often the most convenient, diagnostic radiograph to perform on these individuals. Individuals with an FMD skeletal phenotype together with any of the following: keloid, scoliosis, cervical fusions, cleft lip or palate, or intellectual disability, and in females, deafness, should be screened for mutations in exon 16 of MAP3K7 initially. If the condition is dominantly inherited, and if the craniofacial manifestations are reminiscent of FMD but the radiographs are unremarkable, the 5′ end of MAP3K7 should be examined for pathogenic variants. Another phenotype is associated with missense mutations in this region of the MAP3K7 gene. CSCF is caused by small deletions and missense mutations in MAP3K7 (Le Goff et al., 2016) and is characterized by short stature, brachydactyly, cardiac septal defects and deafness. It is worth noting that the individuals described here often have scoliosis and contractures which sets the diagnosis of AD-FMD apart from CSCF (Le Goff et al., 2016). Therefore, as is the case with FLNA, mutations in MAP3K7 can give rise to an allelic spectrum of disorders.
X-linked-FMD belongs to a spectrum of bone sclerosing conditions, the OPDSDs which includes, listed here in order of increasing phenotypic severity, otopalatodigital syndrome type 1 (OPD1), FMD, OPD2, Melnick-Needles syndrome (MNS), and terminal osseous dysplasia (TOD) (Robertson, 2007). The presentation of the five disorders is distinct in males (Melnick & Needles, 1966; Robertson, 2007). However the severity varies considerably in females, and OPD1, FMD, and OPD2 share overlapping features which can make correct diagnosis difficult (Naudion et al., 2015; Robertson, 2007). The AD form of FMD presents with a more recognisable FMD phenotype in females, the most remarkable feature being supraorbital hyperostosis. There is little to set MAP3K7 c.1454C>T (TAK1, p.Pro485Leu) mutation positive AD-FMD apart from TAB2 mutation positive AD-FMD, however it appears mutations in TAB2, both of which are restricted to exon 5, are comparatively rarely encountered. Missense, presumed loss-of-function mutations in TAB2 have previously been associated with congenital heart defects (Thienpont et al., 2010). The two individuals we have presented here with TAB2 mutations do not have a described cardiac defect. This disparity in phenotype could be explained by the location of the mutations with those conferring a loss-of-function found in the second coding exon of the TAB2 (GenBank: NM_015093.5) and more N-terminal in the TAB2 protein (p.Pro208Ser and p.Gln230Lys). These variants have the same phenotypic outcome as large deletions which encompass the TAB2 locus (Thienpont et al., 2010). In contrast the mutations that cause AD-FMD are located in the third coding exon, resulting in substitutions that are more C-terminal in the protein (p.Gln540Arg and p.Glu569Lys). The number of individuals with TAB2 mutations presented here is small and therefore it is not possible to be certain that TAB2 mutations leading to AD-FMD are not associated with heart defects also.
Both XL-FMD and AD-FMD present with numerous morbidities. Qualitatively the skeletal phenotype progresses in early childhood but there is limited data available on how the condition progresses as affected individuals age. Since the finding that MAPK signaling underpins these phenotypes; evolving complications from progressive skeletal disease could be addressable with agents targeted to this pathway.
ACKNOWLEDGMENTS
The authors would like to thank the participating families and J. Weissenbach for diagnostic expertise. The work was supported by funding from the Marsden Fund and Cure. Kids (S.P.R.). E.W. is supported by an Otago University postgraduate research scholarship.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.