Clinical and molecular characterization of a second family with the 12q14 microdeletion syndrome and review of the literature
Abstract
The 12q14 microdeletion syndrome is a rare condition characterized by low birth weight, failure to thrive, short stature, learning disabilities, and osteopoikilosis. To date, 20 cases of 12q14 deletion have been reported in the literature, displaying both phenotypic than genetic variability. We report on three familial cases, a mother and two brothers, with severe short stature. The mother and elder brother presented with osteopoikilosis while the younger brother had severe short stature and developmental delay. SNP array analysis revealed a 1.9 Mb heterozygous 12q14.2q14.3 deletion in all three patients encompassing 14 genes and 3 miRNAs. In addition, the younger brother carried a paternal 11q13.4 duplication including the SHANK2 gene. This latter patient was investigated for developmental delay and did not show osteopoikilosis, confirming the role of age in the clinical presentation of this condition. To the best of our knowledge, this is the second family described with the syndrome. Comparing the clinical and molecular data of our patients with those previously reported we performed a detailed genotype–phenotype correlation confirming the association between growth retardation and osteopoikilosis when the rearrangement includes both LEMD3 and HMGA2 genes. In addition, we suggest the XPOT, TBK1, WIF1 genes as candidates for the clinical features observed in our patients and discuss for the first time the possible involvement of some microRNAs, when deleted, in the etiology of the phenotypes in 12q14 microdeletion syndrome patients. We expect the interpretation of our findings to be useful both from a molecular point of view and for genetic counseling.
1 INTRODUCTION
The 12q14 microdeletion syndrome was first described by Menten et al. (2007) studying the molecular basis of osteopoikilosis (OPK) (OMIM 166700), an autosomal dominant bone dysplasia characterized by benign, symmetric, and asymptomatic osteosclerosis areas in different parts of the skeleton. In their paper, the authors presented three unrelated patients with, in addition to osteopoikilosis lesions, mild intellectual disability (ID), low birth weight, failure to thrive, and proportional short stature. In each case, a 12q14 heterozygous deletion was detected, ranging from 3.44 to 6.0 Mb in size, with a 3.44 Mb common deleted chromosomal region including LEMD3, HMGA2, and GRIP1 genes, respectively, correlated to familial cases of OPK (Hellemans et al., 2004), human growth regulation (Ligon et al., 2005; Zhou, Benson, Ashar, & Chada, 1995), and ID (Hata, Nakanishi, & Takai, 1998).
Later, Mari et al. (2009) described a young boy, carrier of an overlapping smaller 12q14 deletion, presenting pre- and post-natal growth failure and developmental delay (DD). The 12q14 microdeletion identify did not include LEMD3 and the proband did not show any sign of OPK.
In the same year, Buysse et al. (2009) described two additional patients with 12q14 microdeletion syndrome and growth deficiency, none of them had OPK but showed mild dysmorphic features and DD. In addition, the authors reported a boy with proportionate short stature carrying an intragenic HMGA2 microdeletion useful to confirm the role of this gene in human growth (Ligon et al., 2005; Zhou et al., 1995). Despite variable breakpoints, all the patients described until now shared a critical deleted region of 2.61 Mb in 12q14.3, encompassing 10 RefSeq: WIF1, LEMD3, MSRB3, HMGA2, LLPH, TMBIM4, IRAK3, HELB, GRIP1, and CAND1.
From a clinical point of view the authors proposed that OPK lesions may be absent in infant patients and pre-adolescent age.
One year later, Spengler et al. (2010) described a 16 years old boy with Silver–Russell syndrome (SRS) like phenotype carrying a de novo 12q14 microdeletion, encompassing LEMD3 and HMGA2 genes. In 2011, Lynch et al. (2011) reported two additional cases showing DD, severe growth retardation and a 12q14 microdeletion encompassing HMGA2 and LEMD3.
The first affected family was investigated by Bibb et al. (2011). They reported a 12q14.3 deletion in a mother and her daughter encompassing 14 known genes (including HMGA2, LEMD3, and GRIP1). Short stature and OPK were present in the mother; joint pain and mild ID were present in the daughter.
Afterward, a female child with proportionate short stature, failure to thrive, and speech delay was reported in literature (Alyaqoub et al., 2012). By array-CGH analysis, a 12q14 microdeletion was identified, containing 25 RefSeq genes: the GRIP1 and IRAK3 genes were completely deleted, HMG2A was disrupted, LEMD3 was not included. The specific contribution of the IRAK3 and GRIP1 genes to the microdeletion phenotype, if any, remained unclear.
Takenouchi et al. (2012) described a female patient with a 12q14 microdeletion involving the HMGA2 gene. They suggested the deletion of HMGA2 as contributing to the short stature phenotype and proposed the haploinsufficiency of a gene proximal (centromeric) to HMGA2 as contributing to the macrocephaly phenotype.
More recently (Nso-Roca, Carratalà Marco, Mestre Ricote, & Juste Ruiz, 2014), some authors described another female patient carrier of a 12q14 microdeletion including LEMD3, HMGA2, and GRIP1 genes. Clinical phenotype was confirmed, and further knowledge was added about endocrinological anomalies, recommending the follow up of these patients in pre-pubertal age.
Finally, Mc Cormack et al. (2015) reported two other patients carrying overlapping deletions in 12q14. The deletion in one patient included both the LEMD3 and HMGA2 genes while the deletion identified in the second patient encompassed the LEMD3 gene but not the HMGA2 gene.
Here, we report on the SNP-array results in three familial cases, a mother and two brothers, with severe proportionate short stature. Mother and elder brother presented moreover osteopoikilosis and a 12q14.2q14.3 microdeletion encompassing 17 genes, including LEMD3 and HMGA2. The younger brother carried the same 12q14.2q14.3 maternal microdeletion and a paternal microduplication in 11q13.4 (400 kb in size) including the SHANK2 gene. He had severe short stature and mild DD but did not show any feature of osteopoikilosis.
2 CLINICAL REPORT
The family was composed of the father and mother, and three children.
Patient I:2 (mother of patients II:1 and II:3) was 42-year-old at the time of our first clinical observation, recalled to have been studied during her 1st years of life for failure-to-thrive in “small-for-date syndrome” and to have been tested for GH deficiency (test results not available in 1970). She had never been treated with recombinant growth hormone. When she was 5-year-old, her corresponding bone age was 2-year-old and she showed a mental age of 3 years and 7 months (Stanford–Binet Scale) and an IQ score of 86. At clinical examination, her anthropometric parameters were height 132 cm (−3 SD) and weight 51 kg (+2 SD).
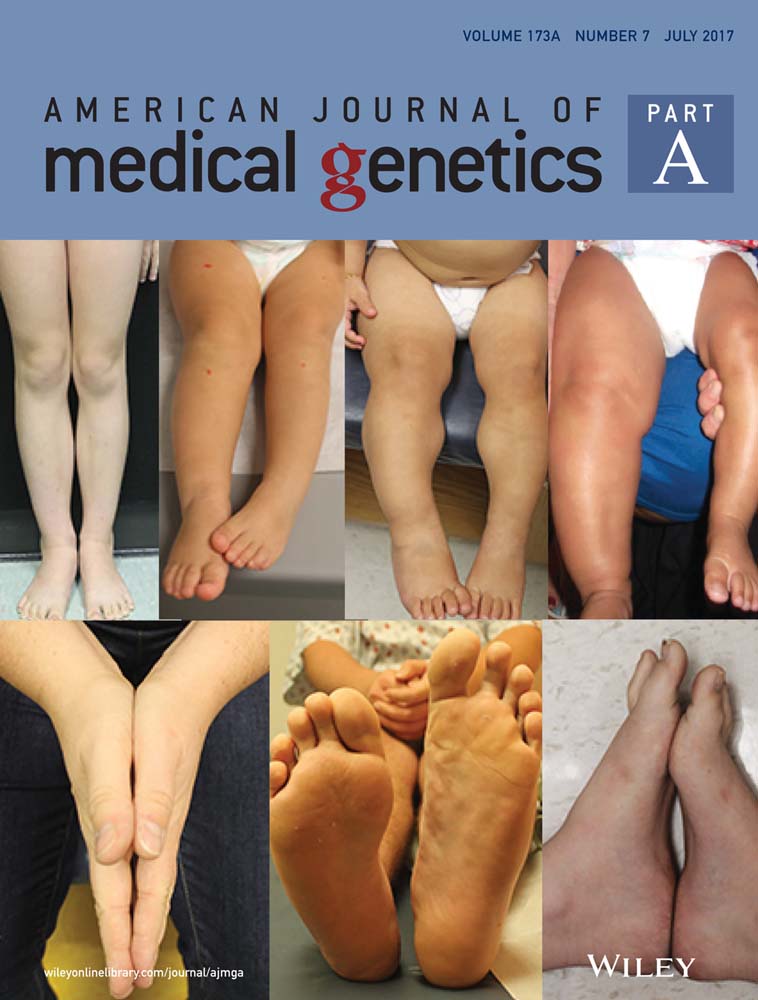
Physical examination revealed triangular face, convergent strabismus, high frontal hairline, thin upper lip, and hypotrophic muscular masses of the upper arms. Full skeleton X-ray showed peculiar signs of osteopoikilosis: multiple osteosklerotic lesions and “spotted bone” images in scapulohumeral and coxofemoral bilateral joints, pelvis, femoral heads, proximal and distal tibial, and radial ephiphysis (Figure 1).

She had attended school without educational supports, had normal intelligence and she had been employed in a factory until now.
Patient II:1 was a boy born at term by Cesarean section, with birth weight of 2,050 g (−3 SD) and length of 40 cm (−3 SD). Ultrasound examination performed at the 7th month of pregnancy showed placental insufficiency and intrauterine growth retardation (IUGR). At the age of 5 months, the patient presented with failure-to-thrive, his weight was 5 kg (−3.64 SD), length was 57 cm (−4.4 SD); when he was 7 months old, bone age delay was present and growth hormone response to glucagon was performed, with a peak GH response of 5.2 ng/ml, and a low IGF-1 value.
Recombinant human growth hormone treatment was started at the dose of 0.025 mg/kg/day, and progressively increased to 0.038 mg/kg/day because of unsatisfying height velocity (7 cm/years in the first 7 years, then 5 cm/years). Treatment was suspended due to advanced bone maturation when the boy was 11 years and 6 months old. We evaluated him for the first time when he was 16 years old: his weight was 52.5 kg (−1.09 SD), height was 141 cm (−4.06 SD), and head circumference (OFC) was 49 cm (−2 SD).
Physical examination revealed dysmorphic features such as triangular face, high anterior hairline, down slanting palpebral fissures, bushy eyebrows, high nasal bridge, and thin upper lip (Figure 2). He had normal intelligence and had attended school without educational supports until that moment.

Routine chemical and hematological exams, standard karyotype, bone and thyroid metabolism, and metabolic screening (plasma aminoacidemia, DMB test, urinary oligosaccharides determination) were normal together with cardiologic, ophthalmologic, ENT (ear, nose, and throat), and abdomen ultrasounds evaluations.
A skeletal survey showed multiple osteopoikilosis lesions in the pelvis (Figure 2), shoulders, wrists, hands and feet, small Schmorl's nodes in dorso-lumbar vertebrae, and on the inner surface of proximal metaphysis of right tibia, an exostosis was present.
On dental radiographs, two secondary teeth were missed with persistent corresponding deciduous ones. Bone densitometry revealed reduced bone calcification.
Patient II:3 was the third son of the sibship, born at term of an uneventful pregnancy by Caesarean section. His birth weight was 1,980 g (−2.38 SD), length was 40 cm (−3.72 SD), and OFC was 30,5 cm (−3 SD). In the first 2 years of life height velocity was 9 cm/year, then decreased to 7 cm/year in his 3rd year of life.
We evaluated him for the first time when he was 3 years and 2 months old, and his height was 81 cm (−4.61 SD), weight was 8,5 kg (−5.96 SD), OFC was 47 cm (−2 SD) . He also showed mild dysmorphic facies characterized by triangular shaped, high nasal bridge, micrognathia, thin upper lip (Figure 3).

We underlined a normal IGF-1 value (100 ng/ml) whereas peak GH response to glucagon was low (3.4 ng/ml). A second growth hormone stimulation test was performed with clonidine (peak GH 6.29 ng/ml). A bone age delay of almost 1 year was present and full skeleton X-ray revealed accentuation of dorsal kyphosis and mild reduction of lower limbs long bones.
Routine chemical and hematological exams, together with standard karyotype, bone and thyroid metabolism, and metabolic screening (plasma aminoacidemia, DMB test, urinary oligosaccharides determination) were normal together with cardiology, ophtalmology, ENT, and abdomen ultrasounds evaluations.
He was found to have delays in fine motor skills. Speech was likewise delayed, with few single words at age 3. GH treatment was started at 5 years of age.
3 METHODS
All reported patients, as well as the father and the normal brother, were analyzed with high resolution SNP Arrays. DNA was extracted from peripheral blood samples using QIAamp DNA Blood Midi Kit (Qiagen, Hilden, Germany). Microarray analysis was performed using the CytoScan HD Array platform and the Chromosome Analysis Suite software v 3.1 (Affymetrix, Santa Clara, CA) according to manufacturer's protocol. Copy number variations (CNVs) were selected by the number of consecutive probes >25 and their size >50 kb. The clinical significance of each CNV detected was assessed by comparison with an internal database of 3,000 clinical samples and public database of CNVs (ISCA, DECIPHER, DGV). All nucleotide positions refer to the Human Genome Feb 2009 assembly (GrCh37/hg19).
4 RESULTS
SNP array analysis revealed a 1.9 Mb heterozygous copy number loss within chromosome region 12q14.2q14.3 encompassing 2,024 oligonucleotide probes in the patients II:1, II:3, and in their mother DNA samples. The proximal breakpoint (centromeric) was located between the last present probe C-6FHUD (64,636,776 bp) and the first deleted probe C-4ZGAL (64,638,433 bp), while the distal breakpoint (telomeric) was located between the last deleted probe C-5WCBQ (66,555,663 bp), and the first present probe C-4TFGK (66,555,756 bp). The molecular karyotype of the patients was arr[hg19]12q14.2q14.3(64,636,776 × 2,64,638,433–66,555,663 × 1,66,555,756 × 2). Moreover, in the patient II:3 SNP array analysis revealed a 0.4 Mb duplication in 11q13.4, inherited from his father, encompassing the SHANK2 gene (data not shown). The proximal breakpoint (centromeric) was located between the last present probe S-3LIPZ (70,433,734 bp) and the first duplicated probe C-3LMHP (70,433,968 bp), while the distal breakpoint (telomeric) was located between the last duplicated probe C-7PZUY (70,842,513 bp) and the first present probe S-4JJHF (70,842,513 bp). Apart from known polymorphisms, no others CNVs were detected.
5 DISCUSSION
In the present study, we describe three familial cases with severe proportionate short stature carrying a heterozygous deletion in the 12q14.2q14.3 region. To the best of our knowledge, this family represents the second case of familiar segregation. A summary of the clinical features of our family and the others cases previously reported in literature carrying a 12q14 overlapping deletion are listed in Table 1, while a molecular comparison is shown in Figure 4.
| Reference | # | Sex | Age at diagnosis | 12q14 deletion size | Inh. | Birth anthropo-metry | Phenotypes | Additional CNVs present in the patient |
|---|---|---|---|---|---|---|---|---|
| Menten et al. (2007) | 3 | F | 16 y | 6 Mb (encompassing LEMD3) | dn | W 2,060 g (<P3) | Failure to thrive; DD; ectopic kidneys; malrotation of the small bowel; synophrys; mild hypertelorism; broad and high nasal bridge; micrognathia and maxillary overbite; OPK | None |
| F | 14 y | 6 Mb (encompassing LEMD3) | dn | W 2,300 g (<P3) | Failure to thrive; DD; Scoliosis; type 1 Arnold–Chiari malformation; reflux nephropathy with small kidneys; mild hypertension; deep-set eyes; bushy eyebrows and thin lips. Diabetes mellitus; OPK; MLR | None | ||
| M | 12 y | 3.44 Mb | dn | At term W at P3 L at P10 | Failure to thrive; DD; yellowish raised areas on the skin overlying the upper chest and flank; triangular face; with widely spaced eyes. Growth hormone levels were normal; normal puberty; OPK | None | ||
| Mari et al. (2009) | 1 | M | 18 m | 1.83 Mb | dn | Pre-term (36 weeks) W 1,730 g (<P10) L 43 cm (−4 SD) OFC 29 cm (−3.66 SD) | Feeding difficulties; DD; Patent ductus arteriosus; ventricular septal defect; triangular face with prominent forehead; down-turned corners of his mouth; a high-vaulted palate; and slight micrognathia. Growth hormone deficiency (growth hormone treatment but after 7 months of treatment the growth velocity did not change) | None |
| Buysse et al. (2009) | 2 | M | 14 y | 8.95 Mb | dn | W 2,550 g (<P5) | Failure to thrive; feeding difficulties; DD; frontal boxing; gastroesophageal reflux. Growth hormone deficiency (growth hormone treatment with mild to moderate response) | None |
| M | 16 y | 3.48 Mb | dn | W 2,055 g (<P3) | Failure to thrive; DD; clustered small nodular elevations of skin; small; pursed mouth with mild a thin upper lip | None | ||
| Spengler et al. (2010) | 1 | F | 1 y9 m | 1.35 Mb (encompassing LEMD3 and HMGA2) | dn | At term W 2,700 g (−1.8 SD) L 46 cm (−2.59 SD) | Feeding difficulties Convex scoliosis; prominent forehead; triangular face; slightly dysplastic ears and clinodactyly | None |
| Lynch et al. (2011) | 2 | F | 7 y | 10.11 Mb | dn | Pre-term (36 weeks) W 2,050 g L 45 cm OFC 32 cm | Failure to thrive; DD; gastroesophageal reflux; convergent squint; high nasal bridge; cupid's bow; clinodactyly. Growth hormone deficiency treated | None |
| F | 3 y | 10.12 Mb | dn | At term W 1,930 g | Failure to thrive; DD; atrial septal defect; hyperactivity; convergent squint; short-broad neck; frontal bossing; hypertelorism; high nasal bridge; short nose; flat philtrum; thin upper lip; micrognathia; low set ears | None | ||
| Bibb et al. (2011) | 2 | F | 12 y | 3.2 Mb | From affected mother | Pre-term (34 weeks) W 1,900 g L 43.2 cm | Failure to thrive; joint pain; DD; hypertelorism; upturned nose; mild micrognathia; clinodactyly; posteriorly rotated ears; hearing lose; white matter loss at magnetic resonance | None |
| F | 45 y | 3.2 Mb | n.r. | At term W 1,900 g (−2 SD) L 40.6 cm (−2.9 SD) | Failure to thrive; short stature; DD; microcephaly; one café-au-lait spot; mild clinodactyly; upslanting palpebral fissures; short fifth metacarpals; curved fingernails; arthritis; carpal tunnel syndrome. Type 2 diabetes; OPK | None | ||
| Alyaqoub et al. (2012) | 1 | F | 7 y | 4.17 Mb | dn | Pre-term (33 weeks) W 1,400 g (<P5) L 40 cm (<P5) | IUGR; feeding difficulties; short stature; DD; Chiari type I malformation; delayed language skills | None |
| Takenouchi et al. (2012) | 1 | 29 m | 4 Mb | dn | Pre-term (27 weeks) W 527 g (−3.78 SD) L 30.5 cm OFC 22 cm | IUGR; short stature; failure to thrive; ASD; cleft lip | None | |
| Nso-Roca et al. (2014) | 1 | F | 7 y | 8.35 Mb | dn | At term W 1,990 g (−2.64 SD) L 44 cm (−2.8 SD) OFC (−0.45 SD) | Feeding difficulties; failure to thrive; DD; gastroesophageal reflux; hypermetropia; triangular face with thin upper lip; prominent forehead; bushy eyebrows with synophrys; yellowish areas on the skin overlying the upper chest and legs; one café-au-lait spot. Growth hormone deficiency; premature adrenarche; precocious puberty | None |
| Mc Cormack et al. (2015) | 2 | M | 3 y | 3.8 Mb | n.d. | At term W 2,550 g (<P3) L 48 cm (P20) | IUGR; severe short stature; relative macrocephaly; mild DD; alternating esotropia; severe myopia; mild ASD | None |
| F | 3 y8 m | 5.6 Mb | dn | Preterm (35 weeks) W 2,600 g (P75) | Absolute macrocephaly; DD; strabismus; prominent brow; large and slightly low set eyes; ASD | None | ||
| Our patients | 3 | M | 16 y | 1.9 Mb | From affected mother | At term W 2,050 g (SGA) L 44 cm (SGA) | IUGR; severe short stature;triangular face; high anterior hairline; high nasal bridge; thin upper lip; GH deficiency with hormone teraphy; OPK | None |
| M | 5 y | 1.9 Mb | From affected mother | At term W 1,980 g (<P3) L 44 cm (<P3) OFC (<P3) | IUGR; severe short stature; DD; triangular face; high anterior hairline; high nasal bridge; thin upper lip; GH deficiency | 0.4 Mb microduplication 11q13.4 inherited from father | ||
| F | 42 y | 1.9 Mb | n.d. | At term SGA | IUGR; feeding difficulties failute to thrive; triangular face; high anterior hairline; thin upper lip; micrognathia; shrilled voice; OPK | None |
- #, patient number; F, female; M, male; y, years; m, months; Inh., inheritance; dn, de novo; n.r., not reported; n.d., not detected; W, weight; P3, third centile; L, length; P10, tenth centile; SD, standard deviation; P20, twentieth centile P5, fifth centile; SGA, small for gestational age; DD, developmental delay; OPK, osteopoikilosis; MLR, melorheostosis; IUGR, intrauterine growth retardation; ASD, autism spectrum disorders; CNVs, copy number variations.

From a clinical point of view our patients, two sibs (II:1 and II:3) and their mother (I:1), have severe proportionate short stature and low birth weight as common phenotypic features.
The older sib (II:1) and his mother (I:1) presented with osteopoikilosis, while the younger sib did not.
Normal intelligence and social performances were present in cases I:1 and II:1, while in case II:3 psychological evaluation revealed speech delay and behavioral anomalies. The 12q14.2q14.3 microdeletion identified encompassed 14 RefSeq genes (C12orf56, XPOT, TBK1, RASSF3, GNS, TBC1D30, FLJ41278, WIF1, LEMD3, MSRB3, RPSAP52, HMGA2, LLPH, and TMBIM4) and three non-coding RNAs (MIR548C, MIR548Z, and MIR6074).
Clinical comparison with patients to date reported is hampered by the fact that all deletions are different in size (from 1.28 to 10.12 Mb) and chromosomal position (Table 1). Nevertheless, all the affected patients to date described exhibit a rather uniform and constant phenotype with low birth weight, failure to thrive in infancy, severe short stature, and neurological disabilities in childhood.
Previous reports indicate that HMGA2 gene is responsible for the aberrant growth pattern observed in these patients. For example, loss-of-function mutation in mice result in the pygmy phenotype, characterized by growth retardation (Zhou et al., 1995). Our data are useful to corroborate the hypothesis that HMGA2 haploinsufficiency is causative for severe pre- and post-natal growth retardation reported in patients with the 12q14 microdeletion.
Some authors suggested as candidate for the neurological features frequently observed in the patients with the 12q14 microdeletion syndrome, the brain-expressed gene GRIP1, which is involved in glutamatergic synaptic transmission (Buysse et al., 2009; Spengler et al., 2010). This gene was not included in the deletion identified in our patients, so we cannot consider it as responsible for the neurological features observed. However, it is located approximately 200 kb telomeric of the deletion identified in our patients so we cannot exclude a positional effect on GRIP1 as already reported and well documented for other genes involved in the etiology of human genetic diseases (Kleinjan & van Heyningen, 1998).
A total of 6/19 cases present with osteopoikilosis lesions. Functional and clinical evidences suggested that LEMD3 gene was correlated with the etiology of osteopoikilosis. In fact, it is involved in bone morphogenetic protein signaling, plays a key role in vertebrate embryonic development (Canalis, Economides, & Gazzerro, 2003) and was previously found as mutated in familial cases of OPK (Hellemans et al., 2004).
Our patients (I:1 and II:1) are useful to confirm that LEMD3 deletion is causative of osteopoikilosis while the absence of this clinical feature in II:3 reinforce the hypothesis that OPK could occur later in life of patients with LEMD3 deletion, in agreement with what previously documented (Bibb, Rosenfeld, & Weaver, 2011; Menten et al., 2007). In light of these evidences, we suggest a careful clinical surveillance in all pediatric and preadolescent age patients with a 12q14 microdeletion encompassing LEMD3.
Moreover, although it is not possible to identify recurrent facial features that make the 12q14 microdeletion syndrome immediately recognizable, some typical dysmorphic features include hypertelorism, frontal bossing, synophrys, micrognathia, triangular face, and clinodactyly not necessarily present in our patients.
In addition to well-established genotype–phenotype correlation we hypothesize that XPOT, TBK1, and WIF1 are good gene candidates for the clinical features observed in these patients.
XPOT encodes a protein belonging to the RAN-GTPase exportin family that mediates export of tRNA from the nucleus to the cytoplasm (Arts, Kuersten, Romby, Ehresmann, & Mattaj, 1998); recent studies highlighted the importance of the nucleo-cytoplasmic transport machinery in genetic diseases and cancer (Leisegang, Martin, Ramírez, & Bohnsack, 2012).
The protein encoded by TBK1 can mediate NFKB activation, in response to certain growth factors, and participates in autophagy pathways. It is plausible to hypothesize that copy number variations of TBK1 could cause a dysregulation either of NF-κB signaling or of autophagy, leading to development of clinical phenotype, as already reported for alterations in genes with similar biological functions (Lee, Hwang, & Lee, 2013).
WIF1 encodes a protein containing a WNT inhibitory factor (WIF) domain, and five epidermal growth factor (EGF)-like domains. It is involved in mesoderm segmentation and its expression is strong during the development of the central nervous system. It is reasonable to assume that WIF1, inhibiting Wnt proteins, play an important role in the development of the central nervous system (Hu et al., 2008). Of note, the deletion identified in our patients encompass also three non-coding RNAs: MIR548C, MIR548Z, and MIR6074. Several reports have indicated a role for non-coding RNAs in the molecular pathogenesis of different developmental disorders (Szulwach, Jin, & Alisch, 2009) and miRNAs copy number change can cause aberrant miRNA expression and/or deregulation of their target genes in patients with neurodevelopmental disorders and congenital abnormalities (Bian and Sun, 2011). In particular, recently, microRNAs have been associated with skeletal and growth defects (de Pontual et al., 2011), ID (Willemsen et al., 2011), and variable expressivity in some syndromes (Ahluwalia, Hariharan, Bargaje, Pillai, & Brahmachari, 2009), all features observed in our patients. Given this evidence, we cannot exclude that deletion of these microRNAs contribute on phenotypes development in 12q14 microdeletion syndromes altering the expression of target genes.
Obviously, more patients and functional studies are needed to confirm a role for these genes and microRNAs in the etiology of the clinical manifestations observed.
Interesting to note, in all the familial cases to date reported, the 12q14 microdeletion has been always inherited from the affected mother. Although the deleted region does not contain known imprinted genes, we cannot exclude the presence of an imprinting mechanism that mediates the expression and the severity of the phenotype. Only the description of additional cases and studies on patients in which the 12q14 microdeletion was inherited from the father, will allow us a better understanding of genotype/phenotype correlation.
Finally, in the younger brother (II:3) we identified an 11q13.4 duplication (0.4 Mb in size), inherited from his father, including the SHANK2 gene. It belongs to the SHANK family genes, which encodes for scaffolding proteins of the postsynaptic density of glutamatergic synapses. CNVs affecting SHANK2 have been reported in patients with autism spectrum disorders (ASD), ID, speech, and developmental delay suggesting a role for the alteration in terms of copy number for this gene in the etiology of these phenotypes (Berkel et al., 2010; Pinto et al., 2010; Schluth-Bolard et al., 2013). Although inherited from an healthy parent, we cannot exclude a contribution for the SHANK2 duplication in the etiology of the observed neurological phonotype in the patient II:3, since that incomplete penetrance and variable expressivity are hallmark of many pathogenic CNVs and occasionally non-manifesting carrier parents have been observed (Ropers and Wienker, 2015).
In conclusion, the description of the clinical and molecular data of our patients allowed us to better characterize the 12q14 microdeletion syndrome as a genomic disorder, which may be recognizable in some cases but must be considered in young patients with low birth weight, height curve below the third centile and familial proportionate short stature.
We confirm that HMGA2 is the main candidate gene for growth retardation since all of our patients had short stature and deletion of HMGA2. In our patients, defective production of growth hormone was proved in both brothers. Thus, we recommend growth hormone (GH) evaluations when the 12q14 microdeletion, involving HMGA2 gene is detected. In the older brother, the stature improved after GH therapy.
Finally, we suggest some other genes, like XPOT, TBK1, and WIF1, as possible candidate genes in the etiology of the phenotypes and, for the first time, the MIR548C, MIR548Z, and MIR6074. Nevertheless, more patients with chromosome aberrations and/or mutations affecting genes of this region need to be reported in order to corroborate our hypothesis, and to establish better genotype–phonotype correlations.
ACKNOWLEDGMENTS
We are grateful to the patient and his family for agreeing to take part in this study. This study was supported by grants of the Italian Ministry of Health (Ricerca Corrente 2016 and RF2011-02350693 to MC).
CONFLICT OF INTEREST
The authors declare that they have no competing interests.