A recessive syndrome of intellectual disability, moderate overgrowth, and renal dysplasia predisposing to Wilms tumor is caused by a mutation in FIBP gene
Abstract
Clinical classification of overgrowth syndromes represents a challenge since a wide spectrum of disorders result in marked overgrowth. Therefore, there is a continuous effort to identify the genetic basis of these disorders that will eventually facilitate their molecular classification. Here, we have identified the genetic etiology and the pathogenetic mechanism underlying a rare autosomal recessive overgrowth syndrome in three affected siblings. The overgrowth phenotype in the patients was accompanied by developmental delay, learning disabilities, and variable congenital abnormalities. To elucidate the genetic etiology of the disorder, whole-genome genotyping and whole-exome sequencing were used. The disease was mapped to 3p21.1-p14.2 and 11q13.1-q13.4, where an in-frame insertion (c.175_176insTAA) in FIBP gene was revealed. The resulting indel (p.H59LN) was predicted to change the protein conformation with likely deleterious effect on its function as one of the fibroblast growth factor signaling mediators. In vitro cellular proliferation assay and in situ hypridization in vivo were then performed to understand the pathophysiology of the disease. The patients’ skin fibroblasts showed an increased proliferation capacity compared to the controls’ explaining the observed overgrowth phenotype. In addition, we detected Fibp expression most notably in the brains of mice embryos suggesting a possible effect on cognitive functions early in development. To date, only one patient has been reported with a homozygous nonsense mutation in FIBP exhibiting an overgrowth syndrome with multiple congenital abnormalities. Taken all together, these findings provide convincing evidence implicating FIBP aberrations in the newly recognized overgrowth syndrome and expand the associated phenotypes to include possible Wilms tumor predisposition. © 2016 Wiley Periodicals, Inc.
INTRODUCTION
Overgrowth syndromes are a group of genetic disorders characterized by excessive growth involving both increased height and weight [Weaver, 1994; Neylon et al., 2012]. In these disorders, all or most parameters of growth and physical development are usually more than two standard deviations above the mean for the patient's age and sex. Some of these syndromes increase the risk of embryonal tumors including Wilms tumor of the kidney [Neylon et al., 2012]. These include Perlman syndrome (PRLMNS, OMIM#267000), Beckwith–Wiedemann syndrome (BWS, OMIM#130650), and Simpson–Golabi–Behmel syndrome (SGBS, OMIM#312870). Infants with BWS are mainly characterized by macrosomia, macroglossia, and abdominal wall defects (including umbilical hernia and exomphalos) with 21% mortality rate [Neylon et al., 2012]. BWS is caused by epigenetic alterations or genomic imbalances at the chromosome 11p15. SGBS infants exhibit pre- and postnatal overgrowth and usually die in-utero [Neylon et al., 2012]. They also have a characteristic face with macrocephaly, ocular hypertelorism, epicanthic folds, broad nasal bridge, macrostomia, and macroglossia. SGBS is caused by mutations or deletions in the GPC3 gene on X chromosome. The prenatal features of PRLMNS include polyhydramnios, fetal macrosomia, prenatal nephromegaly, and fetal ascites [Perlman et al., 1973; Neylon et al., 2012]. The affected neonates have a distinctive facial appearance including a macrocephaly and a prominent forehead, deep-set eyes, a broad flat nasal bridge, an averted V-shape upper lip, and low-set ears. They are abnormally large with enlarged kidneys and liver. Half of the reported cases die in the first few days of life mainly due to renal failure. Survivors with this syndrome typically have developmental delays. PRLMNS is caused by mutations in DIS3L2 gene encoding a cell growth regulator [Astuti et al., 2012].
In this article, we describe a syndrome of moderate overgrowth associated with intellectual disability and Wilms tumor predisposition in multiple affected siblings in a consanguineous family from the United Arab Emirates. This syndrome does not resemble any of the syndromes discussed above (Table I). Our molecular investigations showed that the affected children shared a homozygous region on chromosome 11 where a homozygous insertion in FIBP gene was found in all of them. A recent study found a homozygous nonsense mutation in this gene in a patient from North Africa with overgrowth phenotype and multiple congenital anomalies [Thauvin-Robinet et al., 2016]. Therefore, our results confirm the association of FIBP loss-of-function mutations with a new overgrowth syndrome in humans.
| Clinical features | Makrythanasis et al. [2014] | Thauvin-Robinet et al. [2016] | Present study |
|---|---|---|---|
| General | |||
| Tall stature | + | + | + |
| Developmental delay | + | + | + |
| Learning/intellectual disabilities | +/− | + | + |
| Macrocephaly | − | + | − |
| Predisposition to malignancies | − | − | +/− (Wilms tumor) |
| Benign neutropenia | − | + | + |
| Sensorineural hearing loss | + | − | +/− |
| Dysmorphic features | |||
| Down-slanting palpebral fissures | − | + | − |
| Widely spaced eyes | − | − | + |
| Epicanthic folds | − | − | + |
| Flat mid face | − | − | + |
| Full/thick lips | − | + | + |
| Skeletal abnormalities | |||
| Talipes varus | − | − | +/− |
| Metatarsus varus | − | − | +/− |
| Bowing of tibia | + | − | +/− |
| Spinal abnormalities | + (Scoliosis/exaggerated lumbar lordosis) | − | +/− (Spina bifida L5 in one) |
| Camptodactyly | + (5th fingers) | + (Limitation of thumb extension) | − |
| Cardiac abnormalities | |||
| Ventricular septal defect | − | + | +/− |
| Double chamber right ventricle | − | − | +/− |
| Mitral valve prolaps | − | + | − |
| Ocular abnormalities | |||
| Astigmatism | − | − | +/− |
| Bilateral blue dot cataract | − | − | +/− |
| Retinal coloboma | − | + | − |
| Strabismus | +/− | − | +/− |
| Renal anomalies | |||
| Renal malrotation | − | + | − |
| Left bifid ureter | − | + | − |
| Nephromegaly | − | − | +/− |
| Cystic dysplastic kidney | − | − | +/− |
| Genes | FGFR3 | FIBP | FIBP |
| Mutations | Homozygous c.1637C>A/p.Thr546Lys | Homozygous c.652C>T/p.Gln218* | Homozygous c.175_176insTAA/p.H59LN |
- * Means termination or a stop codon.
METHODS
Participants and Genetic Analysis
A family with three affected children exhibiting a syndrome of neonatal overgrowth and developmental delay was recruited for this study (Fig. 1). Al-Ain District Human Research Ethics Committees approved the study and the family provided a written informed consent for participating in the study.

Whole-genome genotyping of all family members and the subsequent homozygosity mapping analysis was performed using HomozygosityMapper Software (Berlin, Germany) as described previously [Seelow et al., 2009; Akawi et al., 2013] (http://www.homozygositymapper.org). The presence of copy number variations was assessed by Nexus Copy Number software (BioDiscovery Inc., El Segundo, CA). Whole-exome sequencing was performed commercially by Oxford Gene Technology (Oxfordshire, UK). Sanger sequencing was carried out as described previously [Akawi et al., 2013].
Cellular Proliferation Assays
Skin biopsy from patient (II2) and normal controls were sliced into smaller pieces and cultured in 6-well plates in DMEM supplemented with 20% FBS, 2 mM l-glutamine and 100 U/ml Penicillin/Streptomycin at 37°C with 5% CO2. After reaching confluency, cells were trypsinized using 1× of trypsin solution and transferred to T25 (25 cm2) flasks in the same media to be further analyzed and maintained. Fibroblasts from patient (II2) and an unrelated control were cultured in a 96-well plate, 2 × 104 per well, in DMEM with 10% FBS. Cell numbers and the degree of cell proliferation were determined by two assays. The proliferation rates of patients versus controls fibroblasts (five age-matched group of healthy, unrelated controls) were determined by CellTiter-Glo® luminescent cell viability assay following the manufacturer's protocol (Promega, Madison, WI). The luminescent signal was recorded after 48 hr, 10 min after reagent addition using PerkinElmer2030 (PerkinElmer). Experiments were repeated three times, and data represented as the mean of six-plicate wells ± SEM.
The patients’ and controls’ fibroblasts were cultured for 24 hr. Then BrdU (Abcam, San Francisco, CA) incorporation was determined 6 hr later at 450 nm by PerkinElmer 2030 (PerkinElmer, Waltham, MA). Cultured cells without BrdU addition were used as control for non-specific binding. The signal measured in both assays is directly proportional to cellular proliferation rate.
FIBP Tissue Expression Profiling in Transgenic Mice
En1Cre, Fgfr1flox, and Fgfr2flox alleles and their genotyping have been described previously [Kimmel et al., 2000; Trokovic et al., 2003; Yu et al., 2003]. These animals were intercrossed to generate En1Cre/+; Fgfr1flox/flox; Fgfr2flox/flox (Fgfr1cko; Fgfr2cko) embryos. For the wild-type analyses, Cre-negative littermates were used. All mice were maintained in an outbred genetic background (ICR). The national committee of experimental animal research in Finland approved all the animal experiments.
Noon of the day of a vaginal plug was designated as the embryonic day (E) 0.5. Embryos were dissected in sterile PBS, fixed in 4% paraformaldehyde in PBS at room temperature for 2–4 days, changing fresh PFA daily, dehydrated, and embedded in paraffin using an automated tissue processor (Leica, Germany). The samples were sectioned at 5 μm, collected on adhesive Superfrost Plus slides (Thermo Scientific, Waltham, MA), air dried, and stored at +4°C until use. DIG-labeled sense and antisense Fibp cRNA probes were synthetized from a linearized plasmid template (Image ID 6395407; Source Bioscience, Cambridge, UK) according to manufacturer's instructions (Roche, Pleasanton, CA). In situ hybridization was carried out essentially as described elsewhere [Wilkinson and Green, 1990]. The samples were photographed using Olympus BX63 microscope connected to Olympus DP72 camera, and images processed using Adobe Photoshop CS5.
RESULTS
The three affected children in the studied family exhibit mainly intellectual disability, moderate overgrowth, and Wilms tumor as summarized in Table I.
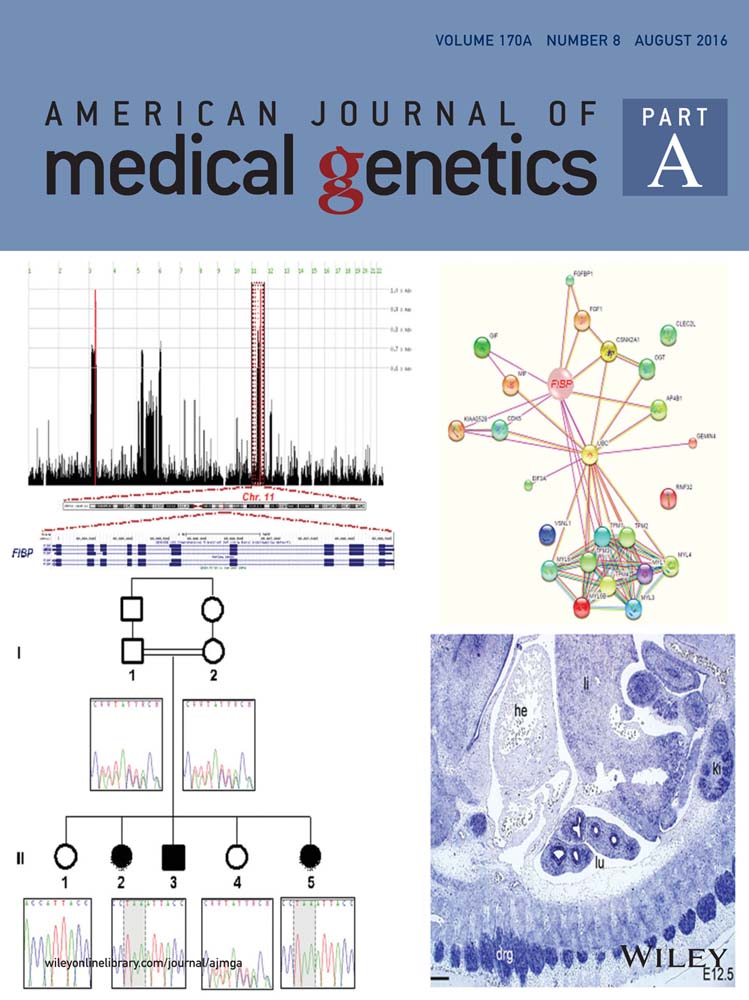
The parents of the affected children are first cousins of Emirati origin (Fig. 1A). They have a total of five children; three of them had distinctive faces, overgrowth in height mainly, developmental delays, learning disabilities, and multiple congenital abnormalities as detailed in Table I. In addition, there is a family history of three miscarriages at 2–3 months gestation and three stillbirths at 6 and 7 months gestation. The father's height was 162 cm and the mother's height was 158 cm, both were of normal intelligence.
Patient 1 (II2) is a 14-year-old girl (Fig. 1B-1, -2), the product of a normal pregnancy and a normal delivery at 39 weeks gestation. Her birth weight was 3,800 gm (90th centile), but no other birth measurements were available. She was noted to have a systolic murmur in the first month of life and evaluation revealed ventricular septal defect (VSD) and a double chamber right ventricle. The VSD was corrected surgically when she was few years old. She presented at the age of 4 years with abdominal distention and on examination a left renal mass was found. Biopsy confirmed the diagnosis of stage-III Wilms tumor. She underwent a course of chemotherapy followed by a left nephrectomy. Postoperatively, a renal ultrasound revealed a simple cyst in the right kidney, which was thought to be of benign. She had delayed developmental milestones and speech. A hearing assessment revealed a mild to moderate sensory-neural hearing loss and, therefore, she required hearing aids. An opthalmological examination revealed astigmatism and bilateral blue dot cataract that was thought to be not significant. She had learning problems but was placed in a normal school with extra help. Her weight at age 14 years was 52 kg (>75th centile, UAE growth charts) and her height was 168 cm (>97th centile, 2.5 SD, UAE growth charts) [Abdulrazzaq et al., 2008]. Her head circumference was 52 cm (50th centile). She had dysmorphic features including a round face with widely spaced eyes, epicanthic folds, and a depressed nasal bridge. She also had a short, small nose with flat mid-face and full lips. She had a persistent neutropenia which was investigated by testing her thyroid-stimulating hormone levels, antinuclear antibodies, and complement component 3/4 activities in addition to examining her bone marrow. All the results were normal, thus a diagnosis of chronic benign neutropenia was made. Comparative genomic hybridization microarray was reported to be normal.
Patient 2 (II3) is the 10-year-old brother of patient 1 (Fig. 1B-3, -4). He was born at 30-week gestation by an emergency lower caesarean due to fetal distress following a normal pregnancy. Birth weight was 1,950 gm (>90th centile). No other birth measurements were available. At birth, he was noted to have severe bilateral talipes equino varus. A left sided convergent squint was noted in the first few months of life and was operated on. His talipes was corrected in the first year of life but assessment at 4 years by an orthopedic surgeon showed that he had severe internal rotation of both femurs and tibias, which required bilateral femoral and tibial derotation osteotomies. He had delayed developmental milestones. At the age of 10 years, his weight was 40 kg (97th centile, UAE growth charts), and his height was 145 cm (>97th centile, UAE growth charts) [Abdulrazzaq et al., 2008]. He had dysmorphic features resembling his sister with similar learning problems. He had some bowing of the legs with metatarsus varus and flat feet. His muscles of the lower limb were weak and wasted, which was attributed to the prolonged use of cast for the correction of his lower limb anomalies. His gait was abnormal with impaired ambulatory capacity. The x-ray of the spine showed spina bifida occulta at L5. His brain MRI and spine MRI (cervical, thoracic, and lumber) were normal. He also had benign chronic neutropenia similar to his sister. An ultrasound of the abdomen revealed normal kidneys.
Patient 3 (II5) is the 3-year-old sister of patients 1 and 2 (Fig. 1B-5, -6). She is the product of a pregnancy complicated by polyhydramnios. Prenatal ultrasound showed cystic dysplastic kidneys. She was born by a lower cesarean. No birth measurements are available. Renal investigations confirmed nephromegaly, bilateral cystic dysplastic kidneys with non-functioning right kidney. At 3 years of age, her weight was 16 kg (97th centile, UAE growth charts) and her height was 110 cm (>97th centile, +2.5 SD, UAE growth charts) [Abdulrazzaq et al., 2008]. She had several dysmorphic features similar to her affected siblings and mild developmental delay.
Genome-Wide Homozygosity Mapping and Whole-Exome Sequencing Identified FIBP Gene as a Disease Candidate Gene
Two homozygous regions were found in the three affected children; these two regions were heterozygous in their parents and the unaffected siblings reaching the maximum homozygosity scores (Fig. 2A). In the affected children, a 6.3 Mb homozygous region was identified on chromosome 3p21.1-p14.2 and an 8.8 Mb genetic interval was shared on chromosome 11q13.1-q13.4 encompassing 214 RefSeq genes; however, none of these are known to cause overgrowth phenotype. There were no potentially pathogenic copy number variations seen in patients. Whole-exome sequencing was carried out on two of the affected children (II2 and II3). A minimum of 79.70% of the on-target regions was covered to a depth of at least 20×. Approximately 47,896 variants from the reference genome were identified of which 10,477 variations were of serious consequences on the corresponding protein products. After filtering out all of the heterozygous reported variations, around 797 variations were left. Within the two disease-candidate regions, only one indel (chr11:65655513:/TTA) was found to be unique and shared between the affected children (Fig. 2B). This was the in-frame insertion of three nucleotides in FIBP gene (NM_198897.1:c.175_176insTAA; p.His59delinsLeuAsn). This indel has not been documented in any of the public databases, in the in-house exomes of individuals exhibiting developmental delays or in the investigated ethnically matched normal controls (Fig. 2C). The indel was found to segregate with the phenotype (Fig. 2C).

FIBP gene encodes a highly conserved acidic fibroblast growth factor (aFGF) intracellular binding protein (FIBP), which is widely expressed in the body proposing it as a housekeeping gene [Kolpakova et al., 2000, 2003, 1998]. Western-blot experiments in humans demonstrated that FIBP is present in two isoforms [Kolpakova et al., 1998]. The mutation in this study caused the deletion of a histidine amino acid number 59 in both isoforms and the insertion of two residues (leucine and asparagine) in its place (Fig. 2D). Although the histidine residue is not highly conserved, it was predicted to change the protein conformation, binding affinity and solvent accessibility by I-TASSER server (http://zhanglab.ccmb.med.umich.edu/I-TASSER/) for protein structure and function prediction. In agreement, PROVEAN (http://provean.jcvi.org/index.php) and SIFT-Indel (http://sift.bii.a-star.edu.sg/www/SIFT_indels2.html) programs predicted this indel to have a deleterious impact on the protein function.
Patients’ Fibroblast Harboring the FIBP Homozygous Mutation Have Enhanced Cellular Proliferation
The mechanisms underlying overgrowth phenotypes were found to be associated with an increased number of cells due to increased cellular proliferation, cellular hypertrophy, or a combination of both [Chung et al., 2012]. Therefore, to understand the mechanism underlying our patients’ overgrowth phenotype and the pathogenicity of the detected mutation, we measured the proliferation rate of the cultured fibroblasts. Interestingly, the patients’ skin cultured fibroblasts showed a notably increased rate of cellular proliferation compared to the controls (Fig. 3A and B). The Thauvin-Robinet et al.'s [2016] study reported similar findings and proposed a similar loss-of-function impact of the indel detected in this study on the FIBP protein. These findings support FIBP's role as a growth regulator.

The Expression Pattern of fibp in Mice Embryos at Different Stages of Embryonic Development Confirms Its Expression in the Central Nervous System at Early Embryogenesis
To explore the pattern of Fibp expression during development, we have used Fibp mRNA-targeted in situ hybridization in mice embryos (Fig. 4a). A uniform weak signal was observed throughout the embryo at embryonic day (E) 10.5 (Fig. 4a-A, -B). Fibp was expressed in all the central nervous system at the E12.5 (Fig. 4a-C, -D). In some areas of the brain a strong signal was seen, in the dorsal mid- and forebrain (Fig. 4a-C). A specific intense signal was noticed in the ventral part of caudal diencephalon (Fig. 4a-C). In the internal organs, Fibp expression was detected in the kidneys and the lungs but not in the heart (Fig. 4a-D). Fibp was expressed in the dorsal root ganglia (Fig. 4a-C). Intensified signal was detected in the caudal part of dorsal mid-brain (inferior colliculi; Fig. 4a-E). In the ventral part of the mid-brain, where there are both dopaminergic and GABAergic neurons, some cells expressed Fibp more strongly than neighboring cells (Fig. 4a-F). However, based on these results alone, it is not possible to determine which cells were expressing the cell-type specificity of Fibp expression. These data confirm FIBP expression during embryogenesis.

To test whether Fibp expression in the brain is regulated during development via some known FGF signaling, we have used Fgfr1 and Fgfr2 conditional double mutant mouse embryos (Fig. 4b). Conditional inactivation of Fgfr1 and Fgfr2 expression in the mid- and hindbrain areas in the E12.5 did not affect Fibp expression. This finding implied that the expression of Fibp in these regions is not regulated via Fgfr1- or Fgfr2-mediated signaling in the brains of mice embryos.
DISCUSSION
A recent study found that a loss-of-function mutation in the FIBP gene underlies an autosomal recessive syndromic overgrowth (mainly in height) associated with macrosomia, learning disabilities, and retinal coloboma in a 17-year-old male (Table I) [Thauvin-Robinet et al., 2016]. This study reports another loss-of-function mutation in FIBP gene causing a remarkably similar syndrome. This syndrome is characterized by an overgrowth and learning disabilities with additional abnormalities including kidney anomalies rendering the patients susceptible to Wilms tumor (Table I). Of the family studied, one of the three affected siblings developed Wilms tumor in early childhood and one has cystic dysplastic kidneys with nephromegaly, however, the third sibling has a normal kidney. This suggests either a clinical heterogeneity of the FIBP mutations or the presence of an additional causative mutation in the other two patients causing their renal anomalies. Therefore, a renal follow-up for patients with FIBP mutations is highly recommended. Table I shows the clinical heterogeneity among the siblings within this family and also compared with the patient from the Thauvin-Robinet et al. [2016] study. Table I also compares the similarities between patients in this study and the patients reported by Makrythanasis et al. [2014]. The affected children in the Makrythanasis et al. [2014] family had similar overgrowth in height associated with intellectual disability and skeletal abnormalities caused by a homozygous loss-of-function mutation in FGFR3 gene. Thauvin-Robinet et al. [2016] proposed that FIBP acts via the FGFR3-mediated pathway, which is critical in signaling cellular proliferation and bone development [Chen and Hristova, 2011].
FIBP was first cloned as a novel protein binding specifically to a member of a family of growth factors called the acidic fibroblast growth factor (aFGF or FGF1) [Kolpakova et al., 1998]. The aFGF functions as a modifier of endothelial cell migration and proliferation, as well as an angiogenic factor. It acts as a mitogen for a variety of mesoderm- and neuroectoderm-derived cells in vitro, thus it is thought to be involved in organogenesis. The binding of FIBP to aFGF was inhibited by heparan sulfate that caused conformational changes in the aFGF [Kolpakova et al., 1998]. It was also demonstrated experimentally that FIBP could bind to the wild-type aFGF but not to the mutated aFGF containing a K118E amino acid change. Thus, the conformation of FIBP protein is important to its function, which we propose to be disrupted by the indel in this report.
K118E-aFGF was able to bind to its cell surface receptors, phosphorylating them, inducing proto-oncogene and mesoderm formation but it was a poor mitogen [Klingenberg et al., 1998; Zakrzewska et al., 2009]. Therefore, the authors had concluded that FIBP might be an intracellular protein involved in the regulation of the aFGF mitogenic activities. Consistently, our results, in addition to Thauvin-Robinet et al. [2016] results endorse the proposed role of FIBP as its loss caused an increased cellular proliferation in patients.
In mice embryos, Fibp was strongly expressed in the developing kidney and neural tissues involved in cognition and sensorimotor functions (including the inferior colliculi; Fig. 4c). This pattern correlates with the defects observed in the patients reported here. However, further in vivo and in vitro experiments are needed to understand the exact mechanistic role of FIBP, as well as its interacting partners and receptors.
To conclude, the present study provides a detailed description of a very rare autosomal recessive overgrowth syndrome with multiple congenital anomalies, expanding its clinical manifestations to include possible cancer predispositions. It also illustrates that the disruption of FIBP and the subsequent increased cellular proliferation is the underlying pathogenetic mechanism. Revealing the genetic and the molecular mechanisms underlying such disorders is crucial for their accurate diagnoses, preventions, and possible treatments [Al-Gazali and Ali, 2010].
ACKNOWLEDGMENTS
We are indebted to the family members for their participation in this research study. We are thankful to Ms. Sania Al-Hamad and Mr. Pramathan T. for their contribution in this study by blood samples collection and DNA extraction. The United Arab Emirates University funds the laboratories of L.A. and B.R.A.