
Current Status of Approaches to Eradicate HIV Infection
Abstract
Direct acting antiretroviral therapy is highly effective in suppressing viremia and preventing progression of human immunodeficiency virus (HIV) to acquired immunodeficiency syndrome (AIDS), but requires strict adherence to lifelong treatment. Upon cessation of therapy, viral rebound is observed within two to four weeks. Recently, significant effort has focused on the development of a finite drug regimen capable of providing sustained virologic response for years or decades; that is, an HIV cure. This review will provide an update on the strategies being pursued and summarize advances in the medicinal chemistry of individual targets.
1 Barriers to HIV Eradication
Direct acting antiretroviral therapy (ART) is highly effective in suppressing viremia and preventing progression of human immunodeficiency virus (HIV) to acquired immunodeficiency syndrome (AIDS), but requires strict adherence to lifelong treatment. In the overwhelming majority of cases, viral rebound is observed two to four weeks after cessation of therapy. Spontaneous drug-free control of HIV is observed in a small number of patients to low levels (long-term nonprogressors) or even below the limit of detection (elite controllers) (1). In one study, a small proportion of patients who started ART during acute infection exhibited delayed time to viral rebound upon treatment interruption (2), as did the few individuals who have undergone allogeneic hematopoietic stem cell transplantation (3). These cases, where a period of treatment leads to viral suppression without the need for chronic therapy, have inspired research toward a pharmacological regimen capable of providing HIV sustained virologic response (SVR) for years or decades, with the aspirational objective of complete eradication of HIV from infected patients.
Two phenomena are likely to prevent ART from providing HIV prolonged SVR, and curative strategies align to address one or both: (i) HIV latency allows replication-competent virus, in a transcriptionally silent state, to persist in host cells and elude detection by host immune responses; and (ii) a subset of infected host cells elude the effects of ART, actively transcribe viral proteins, and produce infectious virus particles without dying in response to viral cytopathic effects or host immune response (Figure 1).

Source: Wolkenberg et al. (4). Reproduced with permission of American Chemical Society.
Strategies to address these two issues are referred to in the literature as “flush” (also “shock” or “kick”) and “kill,” and a consensus has emerged that addressing both, either with a dual-acting single agent or a combination of agents, is likely required to achieve HIV cure (Figure 2) (5).

Source: Wolkenberg et al. (4). Reproduced with permission of American Chemical Society.
A key scientific and ethical challenge for curative therapy is providing acceptable risk/benefit in the context of generally well-tolerated and convenient ART (6). Curative regimens will by necessity be immunomodulatory because cells of the immune system are the target of HIV infection, can play an effector role in killing actively infected cells, and harbor latent HIV provirus; balancing stimulation of these cells (sufficient to reveal latent HIV) with avoidance of broad immune activation (severe safety risk) is a preoccupation across the strategies being pursued (7).
This article is based on a review article published in Medicinal Chemistry Reviews, vol. 51 (2016) (4); it has been expanded considerably and updated to include literature published through 2018. Passages from the original article are reprinted with permission from the American Chemical Society, Medicinal Chemistry Division. To provide an overview of such a broad field, the scope of this review is limited to those studies where molecules and mechanisms were investigated for HIV eradication. Many of these studies were conducted with compounds developed for other indications, most commonly oncology, and detailed SAR for those indications is omitted unless it bears on effectiveness for HIV.
1.1 Latent HIV Reservoir
Latent HIV infection in resting memory T-cells from patients on suppressive ART was first characterized in 1997, when these cells were found capable of producing infectious virions following activation (8-10). Under resting conditions, memory T-cells do not express viral antigens and as a consequence they are not cleared by innate or adaptive immune responses. HIV-infected resting memory T-cells are seeded early in acute infection and appear long lived (t1/2 ∼44 months), indicating that the latent reservoir would take >60 years to decline to basal levels (11, 12), and allowing for the persistence of the virus even if ART is administered. Analysis of T-cell subsets indicates that all types harbor latent HIV infection (13), suggesting that strategies to reactivate latent HIV infection will have to be active in all of these subsets. Recent studies have also shown that homeostatic proliferation due to T-cell receptor agonists or cytokines can contribute to expansion of the latent reservoir in the absence of virus reactivation (14). Cells harboring HIV proviral DNA can be readily detected by sensitive PCR-based techniques, and quantification of the reservoir suggest the reservoir is quite dilute: 1–100 infected cells per million resting memory T-cells (9, 15-18).
1.2 The Persistent Reservoir
Despite the potency of antiretroviral drugs, low level viremia can be detected in the blood of HIV-infected patients on suppressive ART (19-21). The source of this low level viremia is unknown but could represent a lack of sufficient ART penetration to all infected tissues or long-lived HIV-infected cells resistant to the apoptotic effects of HIV infection.
HIV-infected cells can be detected in lymph nodes in ART suppressed patients. T-cell subset analysis has indicated that T-follicular helper cells (TFH), which aid in B-cell maturation and antibody formation in germinal centers, harbor significant infection in this tissue (22, 23). This has two important consequences for HIV pathogenesis. First, CD8 T-cells and NK cells are largely excluded from this site, thus preventing penetration of cells that could clear HIV-infected cells. Second, infection of TFH cells might interfere with B-cell maturation and thereby impact the production of neutralizing antibodies, further blunting the adaptive response to HIV.
1.3 Quantification of the Viral Reservoir
Quantification of the viral reservoir is critical to being able to evaluate curative therapies. Viral reservoir measurements have included quantification of proviral DNA, viral RNA, and viral proteins. There are multiple factors that complicate viral reservoir measurements. First, a majority of HIV proviruses found in infected individuals are defective (24). Defective proviruses may have the ability to produce viral RNA or viral proteins but would not contribute to new infections upon cessation of ART. Quantitative viral outgrowth assays (VOAs) involve reactivation of latently infected cells followed by limiting dilution and coculture with HIV target cells and therefore overcome this obstacle by requiring multiple rounds of replication prior to detection (25). VOA is labor intensive but focused on the replication competent reservoir. In addition to VOA, next generation sequencing technologies have allowed for near full-length provirus sequencing which can separately quantify intact and defective proviruses. A digital droplet PCR assay, the intact proviral DNA assay (IPDA), was recently developed and involves quantification of two amplicons and a probe to enable exclusion of 97% of defective proviruses without sequencing (16). Data from IPDA and near full-length provirus sequencing indicate that the replication competent reservoir is on the order of 100 infected cells per million resting CD4+ T cells in suppressed individuals.
Second, infected cells can exist in both transcriptionally active and silent forms and both contribute to the viral reservoir. Measurements of proviral DNA allow for quantification of both forms; however, many overestimate the size of the reservoir if the methodology does not exclude defective proviruses as described above. VOA and other methods that quantify viral RNA or proteins utilize T-cell activators or latency reversing agents (LRAs) prior to detection in order to get transcriptionally silent proviruses to start producing RNA and proteins. Unfortunately, studies using multiple rounds of activation have demonstrated that not all cells that are capable of producing viral RNA and protein do so upon each round of LRA treatment. This means that RNA- and protein-based methods may underestimate the size of the reservoir and require multiple rounds of latency reversal treatment to best assess reservoir size.
Finally, it is important to note that the assays described above have mostly been used to quantify the reservoir within the periphery. More recently there have been a number of studies performed to assess the reservoir in animal models (26-28). There are also a number of clinical PET imaging studies using antibodies targeting the HIV Env protein that should readout in the near future (29-31). A better understanding of tissue reservoirs and their contribution to the total body burden will greatly impact this field.
2 Latency Disruption
Selective killing of latently infected cells requires a recognition element for the kill effector. Host cell-surface proteins have been investigated for the identification of latently infected cells (32), but to date no single host protein or combination has emerged as a high quality marker. Therefore, LRAs, which induce transcription and translation of viral proteins and thereby flag cells as infected, have been pursued.
When considering LRAs (“flush” agents) there are two important criteria: efficacy (what proportion of the latent cell population is induced), and safety (what level of nonspecific T-cell activation is induced). As HIV transcription likely relies on transcriptional factors that are suppressed in latent memory T-cells, activating HIV transcription in the absence of T-cell activation remains challenging. In general, agents which provide the highest proportion of latent reservoir induction also provide broad T-cell activation, and discerning the balance between these properties is challenging because both vary between assay and model systems; it is not yet clear which models offer the highest predictive value.
2.1 Histone Deacetylase Inhibitors
Histone deacetylase (HDAC) inhibition has been studied extensively as a mechanism of latency disruption, including six published clinical studies (using drugs developed for oncology) and numerous in vitro reports. The 11 human HDAC isozymes catalyze the hydrolysis of acetylated lysine side chains on many cellular substrates, including histones associated with DNA in the form of chromatin. Acetylation of histones, along with methylation and other epigenetic modifications, regulates gene transcription, and inhibition of HDACs stimulates transcription changes across thousands of host genes. HDAC inhibition causes an increase in HIV transcripts in cellular models of HIV latency (33, 34) and in latent cells from HIV positive patients treated ex vivo or in vivo (35-39). Viral protein production in ART-suppressed HIV-infected patients has been observed in response to HDAC inhibitor treatment, although it is not known whether this is sufficient to render cells susceptible to a kill strategy clinically (38, 39). In vitro combination studies in model systems of flush and kill agents have shown proof of concept (40, 41).
In clinical studies, markers of nonspecific T-cell activation are unchanged in response to HDAC inhibition. This important observation in the context of HIV eradication is offset by a number of limiting factors: the proportion of the latent reservoir induced upon HDAC inhibitor treatment may be small (11); most HDAC inhibitor drugs are mutagenic and therefore not suitable for widespread use beyond oncology (42); clinical HDAC inhibitors are associated with a common set of toxicities (hematological changes, nausea, fatigue) which appear invariant even as HDAC isozyme selectivity is narrowed, and these toxicities may limit the use of HDAC inhibitor drugs among HIV patients (43). An additional limitation has emerged in recent in vitro studies of HDAC inhibitor effects on HIV-specific CD8+ T-cells. These CD8+ T-cells represent an important endogenous kill effector which is envisioned to complement flush by LRAs. Multiple studies provide evidence that some HDACi may impair CD8+ T-cell function thereby diminishing their potential to drive reservoir reduction via flush and kill. However, effects were different among vorinostat (1), panobinostat (2), and romidepsin (3), with panobinostat and romidepsin exhibiting the most profound effects on markers of T-cell function. Whether this difference arises from individual inhibitor potency, isozyme selectivity, dose interval, or HDAC inhibition kinetics remains to be determined (40, 44-47).
The HIV latency reversal clinical studies with vorinostat (35-37, 48), panobinostat (38), and romidepsin (39, 49, 50) report similar results. Increases in cell-associated HIV mRNA in T-cells, a marker of HIV transcription, coincided with peak HDAC inhibition as measured by acetylated histone accumulation. Panobinostat and romidepsin also increased viral loads, indicating that HDACis could actively flush latent cells. An intriguing aspect of these studies is that elevations in cell-associated HIV mRNA persist after the treatment period, in some cases for weeks. Mathematical modeling of these results has suggested that this is due to the induction of a quasi-activated T-cell memory state, which is more permissive of HIV transcription in resting cells (51). However, despite consistent evidence for latency reversal, none of the studies indicates a change in the size of the latent reservoir; that is, the HDAC inhibitors alone provide flush but not kill. One clinical flush and kill combination study of romidepsin co-dosed with a p24-based HIV therapeutic vaccine and recombinant human granulocyte macrophage colony stimulating factor has been reported. An effect on reservoir size was observed, though the magnitude of the reduction is small and ART analytical treatment interruption showed no prolongation in the time to viral rebound (49) (Figure 3).

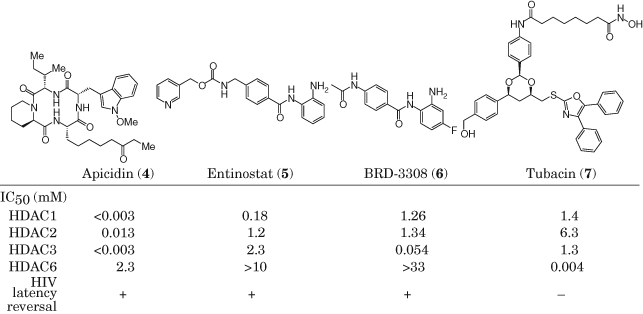
Clinical HDACis are approved for use in oncology and come with a variety of adverse effects that would not be readily acceptable in the current state of HIV care. Therefore, mechanism of action studies has been performed to determine if inhibitors can be designed with a focus on HIV reactivation. Vorinostat, panobinostat, and romidepsin inhibit HDACs with low selectivity, and emerging evidence suggests that inhibition of a subset of isozymes, in particular the Class I HDACs (isozymes 1/2/3), is sufficient to reverse HIV latency (52, 53). For example, inhibitors analogous to apicidin (4) that inhibit HDACs 1/2/3 or 1/2/3/6 were observed to reverse latency in both a Jurkat model and in cells from HIV+ patients. However, inhibition of HDACs 1/2 or 6 alone (as with Tubacin, 7) was insufficient for latency reversal, leading to the conclusion that HDACs 1/2/3 are required. Separate studies with HDAC1/2/3-selective inhibitor entinostat (54) (5) and HDAC3-selective inhibitor BRD-3308 (53) (6), which integrate results from selective siRNA knockdown of particular HDAC isozymes, reach similar conclusions (Table 1).
 |
The medicinal chemistry of HDAC inhibitors for oncology, including modulation of selectivity profile, spans three decades and is beyond the scope of this review. The reader is directed to several reviews (55-59).
2.2 Epigenetic Regulators Beyond HDAC Inhibitors
The encouraging clinical results with HDAC inhibitors have demonstrated that transcriptional regulation of HIV is likely connected to the chromatin state of the HIV genome (60). As HIV-1 integrates into the host chromatin, the location of the integrated viral genome and the epigenetic modifications at the proviral site are involved in the preservation of HIV-1 latency (61). Therefore, additional epigenetic mechanisms for modulating the transcriptional accessibility of the HIV LTR also hold promise as HIV flush agents.
Bromodomain and extra-terminal domain inhibitors (BETis) have been identified as a new class of LRAs (62). Several studies have shown that BRD4 inhibitor JQ-1 (8) can reverse HIV-1 latency alone ex vivo and synergistically with PKC agonists (63-65). However, JQ1 exerted severe dose-limiting cytotoxicity (11). Medicinal chemistry efforts to improve JQ-1 using fluorous-tagged multicomponent reactions led to the identification of UMB-136 (9), a dimethylisoxazole BETi carrying an imidazopyrazine scaffold (66). This second-generation BRD4 inhibitor increased viral production through the release of P-TEFb, and reactivated HIV-1 in multiple cell models of HIV-1 latency with better efficiency than JQ1, and more importantly, enhanced the latency-reversing effects of PKC agonists (67) (Figure 4).

Among other epigenetic modifying agents, DNA and protein methyltransferases (PTMs) and demethylases are also involved in HIV infection and essential for HIV-1 transcription. In this context, DNA methylation inhibitors (DNMTis) are considered latency reversing agents. This category includes FDA approved decitabine (5-aza-2-deoxycytidine, 10), which was shown to induce HIV-1 viral gene expression in cell line models, alone (68) and in combination with prostratin (69). On the other hand, decitabine demonstrated only limited reactivation in aviremic patient derived cells (70). Therefore, the role of DNA methylation in HIV-1 is not clear and still debated in the field (Figure 5).

Likewise, additional epigenetic regulators such as PTMs and demethylases have emerged as new targets in HIV-1 infection. Results with histone lysine methyltransferases (HKMTs) as LRAs are somewhat conflicting. While GLP and G9a HKMT inhibitor BIX-01294 (11) (61, 71, 72) and EZH2 HMKT inhibitor 3-deazaneplanocin A (12) clearly show latency reversing activity (73), dual EZH1/EZH2 inhibitor GSK-343 (13) only sensitized resting cells to reactivation by the HDAC inhibitor vorinostat and does not work alone (74). More recent studies, however, show that inhibition of EZH2 with GSK-343 or EPZ-6438 (14) can induce the reactivation of latent proviruses. The different observations in these studies could be due to the different cell lines and experimental conditions between experiments (75) (Figure 6).

The SUV39H1 histone methyltransferase inhibitor chaetocin (15) increased induction of latent HIV-1 expression in association with loss of H3K9 trimethylation at the LTR promoter, and a corresponding increase in H3K9 acetylation. In addition, minimal toxicity and no T-cell activation was observed (76). Interestingly, chaetocin reactivated HIV-1 from latency in CD4+ T-cell cultures isolated from ART-patients, and was synergistic with HDACis (71). Chromatin immunoprecipitation studies revealed that the proviral LTR is highly enriched with euchromatic histone–lysine N-methyltransferase 2 (EHMT2) and displacement occurs rapidly upon proviral reactivation. EHMT2 inhibitor UNC-0638 (16) demonstrated reactivation of latent proviruses in cells derived from ART suppressed HIV-1-infected individuals as a single agent and was synergistic with vorinostat (75).
A small hairpin RNA (shRNA) screen in HIV-infected T-cell lines identified the human lysine methyltransferase SET and MYND domain-containing protein 2 (SMYD2) as a new potential target for HIV-1 latency reversal. This enzyme was validated by knockdown studies and pharmacological inhibition using SMYD2 inhibitor AZ-391 (17). When AZ-391 was tested in J-Lat cell lines in combination with previously known LRAs, synergy was observed with JQ1 and vorinostat, but was not even additive with ingenol-5,20-dibenzoate. When CD4+ T-cells from HIV-1-infected individuals on suppressive ART were treated with AZ-391 alone or in combination with JQ1, increased HIV-1 mRNA levels were observed for both drugs alone but in the absence of any additive or synergistic effects (Figure 7) (77).

Although research into epigenetics beyond HDAC inhibitors for HIV latency reversal is less advanced, some methyltransferase inhibitors are in clinical trials for oncology indications, making future clinical use in HIV infection more feasible. This class of inhibitors has also demonstrated synergy with other classes of latency reversal agents which could pave the way for possible combination treatments. Such effort will require development of a more thorough understanding of the safety liabilities of these mechanisms in the HIV patient population.
2.3 Protein Kinase C Agonists
Protein kinase C (PKC) agonists exhibit impressive potency in multiple in vitro models which examine reactivation of latent HIV. PKC mediates many cellular activities including cell growth and cell death (78). Latent virus reactivation is mediated via agonism of PKC, which triggers the NF-κB pathway with subsequent modulation of regulatory elements on the HIV long terminal repeat promoter. The PKC agonists examined thus far have primarily been derivatives of natural products, including ingenol derivatives (18), phorbol esters (19), bryostatin (20), and glycerol esters (64, 65, 79).
The diterpene ingenol is a secondary metabolite of Euphorbiacea plant and its derivatives inhibit HIV reverse transcription by activating PKC (80, 81). Some semisynthetic analogues such as ingenol 3-angelate and ingenol 3,20-dibenzoate are PKC agonists with antitumor and thrombopoietic activities, respectively (82, 83). Attempts to synthesize less toxic compounds retaining their HIV latency reverse activities led to new C3-ingenol ester derivatives: trans-cinnamate (ING A, 18a), caprate (ING B, 18b), and myristate (ING C, 18c) (84). In addition, ingenol-3-angelate (PEP005, 18d), an FDA approved anticancer drug from the ingenol family, was found to effectively reactivate HIV-1 from latency in primary CD4+ T-cells from HIV-infected individuals receiving ART (85).
Ingenol B is one of the more advanced PKC agonists promoting PKC activation and NF-κB nuclear translocation. It has been studied in several in vitro HIV-1 latency models and evaluated ex vivo in rCD4s isolated from suppressed SIV-infected macaques. In all cases, the compound significantly increased viral transcription (86, 87). More recently, ingenol B was tested in combination with HDACi vorinostat in a SIV-infected macaque model to evaluate the contribution of the brain to the latent reservoir and reactivation during ART. This study led to an increase in viral load in cerebrospinal fluid (CSF) indicating that the CNS harbors latent SIV genomes, which represents the first in vivo proof of concept that brain is a potential viral reservoir to consider during HIV cure strategies (88). While not applied to HIV reactivation, use of selective CH oxidation methodology with ingenol demonstrates that new chemistry approaches can enable modulation of PKC isozyme activity (89) (Figure 8).

Phorbol esters, including phorbol 12-myristate-13-acetate (PMA, 19) and prostratin (20), are natural products with strong PKC activation activity. PMA is often used in vitro in combination with the potent Ca2+ ionophore ionomycin as a strong inducer of T-cell activation and latent proviral transcription (11, 90). Use of PMA is limited to in vitro studies due to its tumorigenic properties (91). Prostratin is a natural product originally isolated in the 1970s (92). Its therapeutic potential was identified in the early 1990s as the main active component of the Samoan medicinal plant Homalanthus nutans (93). Preliminary studies demonstrated that prostratin promoted translocation of NF-κB into the nucleus and subsequent binding to LTR in Jurkat-derived latent T-cell lines to induce HIV-1 reactivation (94). Prostratin is not tumorigenic, possessing instead selective inhibitory activity against certain cancer cell lines (95). A novel series of prostratin C13 analogs exhibited superior activity over prostratin in a latently infected model cell line. Analog 21 was also found to potently induce the expression of latent virus from PBMCs isolated from ART suppressed HIV-infected individuals at concentrations in which prostratin is inactive (96) (Figure 9).

Bryostatin-1 (22) is a PKC agonist derived from marine invertebrate with well-characterized antineoplastic activity in vitro (97, 98). While bryostatin-1 demonstrates activity in latency models in vitro, it did not impact HIV transcription in a clinical trial by measuring cell-associated viral RNA; however, no evidence for PKC activation was observed, likely due to low plasma levels (99). In addition to flush activity, bryostatin-1 has also been shown to inhibit acute viral replication through down regulation of the CD4 receptor, which could supplement ART in the flush and kill approach to protect uninfected cells (100).
In general, studies in several primary CD4+ T-cell models of HIV-1 latency have corroborated the potential of certain PKC agonists as potent latency reversal agents (11). Nonetheless, PKC agonists also induce mechanism-dependent cytokine release and are potentially tumor-promoting. The design of PKC isozyme-specific molecules will facilitate a more detailed understanding of the factors that control potency and toxicity; i.e. latency reactivation vs. cytokine generation. Towards that end, gnidimacrin (23) has been shown to activate latent HIV through selective activation of the PKC β pathway (101) (Figure 10).

Interestingly, these agents synergize with other classes of LRAs, such as HDACis to induce potent reactivation of latent HIV-1 (102). However, PKC modulators induce a much stronger T-cell activation compared to HDACis, coupled with nonspecific cytokine production and T-cell proliferation (103).
2.4 BIRC2 Inhibitors
Activation of the canonical NF-κB pathway, as highlighted by PKC agonists, is a powerful way for activating latent HIV but elicits a strong, potentially unmanageable cytokine response. On the other hand, the activation of the noncanonical NF-κB pathway is a slower process with a long-lasting response and higher functional selectivity (104). These features provide a potential mechanism for separating potency from toxicity and render the noncanonical NF-κB pathway a suitable target for HIV-1 reverse transcription.
A siRNA screen led to the identification of the ubiquitin ligase BIRC2/cIAP1 as a repressor of the noncanonical NF-κB pathway. In HIV-infected cells, BIRC2/cIAP1 was found to be a negative regulator of LTR-dependent HIV-1 transcription, with SMAC (second mitochondrial-derived activator of caspases) mimetics such as 24 able to inhibit this pathway and activate latent HIV (105). AZD-5582 (25) (106), is an anticancer clinical candidate SMAC mimetic reported to function as a potent LRA in vitro and ex vivo with limited toxicity in CD4+ T-cells. Treatment of CD4+ T-cells from ART-suppressed, HIV-infected patients with AZD-5582 led to significant increase in cell-associated full-length HIV RNA (107) (Figure 11).

SMAC mimetics have been studied in combination with other LRAs and in vitro in resting CD4+ T-cells isolated from ART-suppressed patients demonstrated potent synergistic activation of the latent reservoir (105). In this context, birinapant (26) (108) showed only a slight increase of expression of latent HIV-1 infected cells when administrated alone, while in combination with PKC activator PEP005 (18d) it induced a strong upregulation of transcription, as well as enhanced apoptosis of latent HIV-1 infected cells (109). Moreover, recent studies have found that SMAC mimetics (birinapant 26, GDC-0152 27, and embelin 28) can selectively kill long-lived resting memory CD4+ T cells without increasing virus production promoted by autophagy and apoptotic mechanisms (110, 111). These results suggest a potential treatment option to decrease HIV-1 reservoirs by providing both flush and kill in one molecule (Figure 12).

2.5 Toll-Like Receptor Activators
Toll-like receptors (TLR) reside on the surface and in intracellular compartments of adaptive and innate immune cells and serve to recognize a variety of antigens. Upon activation, multiple pathways are stimulated including the transcription factor NF-κB. TLR agonists have been shown to activate latent HIV reservoirs in vitro through this NF-κB pathway (112, 113). In addition to activating transcription, TLR activation is also thought to activate immune system-mediated clearing of HIV-infected cells; clinical evidence for this effect includes viral DNA reduction with a single-stranded DNA, CpG-derived agonist of TLR9 (112). Significant toxicity was reported for the CpG-derived agonist, and an alternative DNA construct MGN1703 was subsequently developed (114). Unlike the CpG oligonucleotide, MGN1703 lacks modified nucleotides and exhibits an improved safety profile. A small clinical study of MGN1703 in HIV-1 infected individuals was performed (115): activation of plasmacytoid dendritic cells was observed, along with increases in plasma IFN-α2 and IFN-stimulated genes in CD4+ T-cell populations. Increases in HIV plasma RNA levels were reported.
TLR7 has also been pursued for HIV latency reversal, and a small molecule agonist (GS-9620, 29) was shown to activate latent HIV in vitro (116). Interestingly, this compound also inhibits HIV infection (117). In separate studies, GS-9620 and a structurally related TLR7 agonist, GS-986, reduced SIV DNA in infected monkeys suggesting both flush and kill effects, although it is not known to what extent these effects were due to the stimulation of cytokines by this mechanism (118). Combination of GS-986 with the therapeutic vaccine Ad26/MVA in the SIV-infected rhesus macaques was effective at decreasing viral DNA, improving virologic control, and delaying rebound after treatment interruption (119). A clinical trial evaluating the impact of TLR7 agonism on HIV viral reservoir is underway (Figure 13).

2.6 Screening Approaches to Identify Novel Agents/Mechanisms
High throughput screening offers a way to identify and prioritize new molecules for HIV latency reversal regardless of their mechanism. For example, disulfiram, an aldehyde dehydrogenase inhibitor, was identified through unbiased screening in a primary T-cell model of latency (120) and has been evaluated in clinical studies (121). Natural product libraries also serve as a source for new chemical matter as highlighted by the identification of radicicol (28), pochonin B (29), and abyssomicin 2 (30), which reverse latency via uncharacterized mechanisms (122, 123). Screening in a primary CD4+ T-cell model identified benzotriazoles as LRAs which cause accumulation of phosphorylated STAT5 at the HIV LTR via inhibition of STAT5 SUMOylation (124). Screening has also been used to identify molecules which can potentially synergize with known reactivation agents, a strategy which aims for the reduction in effective dose of both agents, potentially improving safety margins (125, 126). While screening is powerful, this approach by necessity must use model systems for HIV latency; as a consequence, any result from a screen must be validated in patient derived cells from multiple donors (Figure 14).

3 Elimination of Infected Cells
3.1 Proapoptotic Agents
Several approaches to eliminate HIV-infected cells target existing host mechanisms that regulate cell death. Among the cell-death mechanisms, apoptosis is highly regulated and well understood (127). It is comprised of an intrinsic pathway, responding to intracellular stress signals, and an extrinsic pathway, governed by extracellular factors. As the host relies on apoptosis to clear pathogens, the virus manipulates apoptotic pathways to optimize its survival. Several agents are known and have been tested to treat HIV infection. Venetoclax (Bcl-2 antagonist), Acitrecin (Rig-I agonist), and Birinapant (SMAC mimetic) have been tested alone or in combination approaches with existing immunotherapies for their effectiveness in accelerating clearance of HIV+ cells (128).
A recent publication (129) describes a novel strategy toward selective induction of apoptosis in HIV+ cells. In this approach, virion budding can be prevented by leveraging the ability of small molecule L-HIPPO (31) to block Pr55Gag viral protein from translocating to the plasma membrane. Blocking Pr55Gag results in the accumulation of viral proteins within the cytoplasm and consequent premature death of the host infected cell (Figure 15).

3.2 Antibody and Antibody-Derived Molecules
Following the reactivation of latent HIV, viral antigens are presented on the surface of the cell and thus can be targeted by antibodies or antibody-derived molecules.
3.2.1 Immunotoxins
Immunotoxins are chimeric proteins comprised of a targeting domain (directed to a specific cell-surface epitope) and a toxin effector domain derived from a plant, bacterial, or animal source (130). HIV specificity is most commonly achieved by targeting several HIV Env-binding domains (131-135). Although initial clinical trials using immunotoxins in HIV-infected individuals failed to have sustained impact on immunological or clinical markers (136), some immunotoxins have been reported to reduce levels of HIV-infected cells that persist in a humanized mouse model (137). More recently, data has been published describing the performance of immunotoxins in the SHIV-infected macaque. While the tested immunotoxins were well tolerated in this model, efficacy was compromised by immunogenicity against the immunotoxin itself, which remains a problem to be solved for this strategy to become viable (138).
3.2.2 Bispecific Antibodies
Bispecific antibodies have been designed to target cytotoxic T-cells to multiple tumors and viral infections (139-144). Bispecific antibodies bridge CD8+ T-cells to target cells via an anti-CD3 antibody arm (CD8 cells) and an antibody raised to a cancer antigen (target cells), bypassing the requirement for T-cell receptor engagement (74, 145, 146). In addition to CD3, NK-targeting bispecific antibodies have also been published (147, 148).
Bispecific antibodies have been produced that utilize an antibody specific for HIV Env proteins on infected cells and have been demonstrated to induce selective CD8 cell mediated killing of HIV-infected T-cells in vitro. Importantly they can also induce killing of latently infected cells treated with LRAs. As persistently infected cells exist in germinal centers of lymph nodes, it will be important to ascertain whether the effector cells can penetrate into these areas during treatment (Figure 16).

Source: Wolkenberg et al. (4). Reproduced with permission of American Chemical Society.
Recently, the bispecific T cell-engaging (BiTE™) technology, originally developed to treat cancer, has been applied to HIV. This construct is engineered from two linked single-chain antibodies which are specific to antigens presented on the targeted cell and to CD3 (149). BiTE constructs demonstrated efficient in vitro killing of gp120-expressing cells (150, 151). In oncology, BiTE constructs have demonstrated superior efficacy and lower immune activation than traditional bispecific antibodies. This is likely due to their smaller size, able to bring effector and target cells in closer proximity, and their monovalent engagement of the TCR complex on CD3 (152). BiTEs are therefore an attractive alternative to bispecific antibodies for the treatment of HIV infection. For comprehensive reviews on bispecific antibodies, see Refs (153-155).
3.2.3 Broadly Neutralizing Antibodies (bNAbs)
Broadly neutralizing antibodies (bNAbs) are historically known as a class of antibodies able to directly block the biological activity of an antigen without the direct help of the immune system (156-164). Several very potent bNAbs against various strains of HIV have been identified, targeting a variety of epitopes. These bNAbs have been studied for their ability to suppress viral load and sustain viremic control after termination of ART. In 2013, Horowitz showed that bNAbs are able to fully suppress viremia in HIV-infected humanized mice; however, viral rebound was observed in all mice within 12 weeks from suspension of treatment (165). Following this work, bNAbs in combination with flush agents demonstrated a decreased rebound rate in humanized mice (166). More recently, there have been a number of studies looking at bNAbs in the clinic. Schoofs et al. (167) demonstrated that treatment with a bNAb (3BNC117) improved host humoral immunity to HIV-1 indicating that these antibodies could do more than just neutralizing existing virus. These data suggest that bNAbs could play a role in boosting the immune system in preparation for treatment interruption.
3.2.4 Antibody-recruiting Molecules (ARMs)
Antibody-recruiting molecules (ARMs) have been described which link a small molecule gp120 ligand with a dinitrophenyl group (168). The former localizes ARMs on the surface of HIV particles as well as cells actively producing virions while the latter binds anti-dinitrophenyl antibodies, which are present in high concentrations in the blood of most humans. The complex formed between anti-dinitrophenyl antibodies, ARMs, and gp120-expressing cells was shown to mediate selective, complement-dependent killing of HIV-infected cells. In addition, the ARMs inhibited the entry of live HIV virus into human T-cells (169, 170). Another approach uses CCR5 antagonist Maraviroc as the targeting moiety (171).
3.3 CAR-T-based Approaches
Data from long-term nonprogressors and elite controllers have demonstrated that CD8+ T-cells can play an important role in maintaining immune control over HIV. To this end, novel strategies are being employed to enhance CD8+ T-cell responses in HIV-infected patients. One such method is the infusion (via adoptive cell transfer) of engineered CD8+ T-cells that express a chimeric antigen receptor (CAR-T cells) (172) directed against HIV (173). The engineered T-cell receptors are able to target HIV-infected cells, most often through recognition of HIV Env, and allow for these modified patient cells to mediate CD8 directed kill. One issue with this method is the nonspecific activation of cytokines (predominantly IFN-g and IL-6) that can induce significant side effects.
Ex vivo expanded HIV-1 multi-antigen specific T-cell therapy (HST-NEET) is currently being investigated clinically as a therapeutic strategy in HIV-infected individuals on ART (174). This new and ambitious strategy aims to improve on CAR-T therapy using hematopoietic stem/progenitor cells (HPSC), where HPSC are engineered to carry the HIV-targeting CAR genes. HPSCs are regenerative, so that the host can potentially maintain sufficient levels of the HIV-recognizing receptor over time. In vivo experiments on SHIV-infected primates demonstrate this concept, with the modified cells being produced for more than two years without significant adverse events (175). For a review on CAR-T treatment of HIV, see Ref. (176).
3.4 Immune Checkpoints
Another strategy that can be employed for the clearance of HIV+ cells is to target immune checkpoints, which are regulators of immune activation that are expressed on immune cells and trigger immunosuppressive pathways. Modulation of checkpoints is important for the virus to allow constant infection while preventing overinfection, and interfering with checkpoint blockade has the potential to restore T-cell function. Antibodies against several immune checkpoint proteins are known, particularly for the treatment of various cancers. For example, antibodies targeting PD-1 (pembrolizumab; nivolumab; cemiplimab), CTLA4 (ipilimumab), and PD-L1 (atezolizumab; avelumab; durvalumab) are all on the market and/or in advanced clinical trials for oncology. One factor to consider for HIV treatment is that different checkpoint proteins are predominant in different cell populations.
Blockade of the inhibitory PD-1/-L1 interaction to restore CD8 T-cell function has been explored in rhesus models of HIV. Promising results were obtained with antibodies to both PD-1 and PD-L1. The anti-PD-1 antibody study (177) was performed in the absence of ART, and rapid expansion of virus-specific T-cells with enhanced function was noted in blood and gut tissue. Significant (2- to 10-fold) reductions in viral load were observed and persisted several weeks after blockade, resulting eventually in improved survival versus control antibody. In a separate study (178), ART-suppressed rhesus macaques were treated with the anti-PD-L1 antibody BMS-936559 and assessed after ART interruption. A subset of treated animals exhibited significantly reduced viral load post-treatment interruption, and the same animals showed stable SIV DNA levels as a measure of reservoir size.
More recent work on PD and PD-L1 blocking in SIV-infected macaque on ART has shown more limited expansion in CD8+ T-cells (179), raising the question of whether the significant presence of antigen is required for a functional response. Similarly, studies with anti-CTLA4 antibodies on SIV-infected macaques on ART have shown modest increase in both HIV-specific CD4+ and CD8+ T-cells in lymph nodes (180).
LAG3 and TGIT are immune checkpoint proteins that have shown higher levels of T-cell expression in HIV+ patients compared to negative controls (181, 182). In vitro studies show that blocking TGIT and PD-1 results in CD4+ activity enhancement (183), suggesting that they may offer potential new targets for HIV treatment.
Initial clinical data to assess PD-1/-L1 blockade for HIV treatment on ART-suppressed individuals indicates some enhancement of HIV-1 specific immunity in a subset of participants; (184) however, the study was ultimately stopped due to retinal toxicity observed in a macaque study that was run at the same time. New clinical trials have recently been announced to evaluate the safety and immunotherapeutic activity of anti-PD-1 antibodies cemiplimab (185) and pembrolizumab (186) in HIV-1 infected participants on ART without cancer. Several other studies are ongoing on HIV+ cancer patients to assess checkpoint blockade effects on both HIV and tumor suppression (187-189). For a recent review on immune checkpoints and HIV, see Ref. (190).
3.5 Therapeutic Vaccine Strategy
The goal of therapeutic vaccination for HIV is to boost the immune system so that it is better able to target and kill infected cells rather than prevent initial infection. There are numerous ongoing studies of therapeutic vaccines for HIV utilizing modalities including DNA, RNA, peptides, proteins, and viral vectors (191-193). While many of these approaches effectively generate immunogenicity the keys to success in this area are to decrease the viral reservoir and elicit a change in viral rebound after discontinuation of ART. A recent clinical study with Vacc-4x, a peptide vaccine which includes four modified peptides corresponding to conserved regions of HIV p24, in combination with the LRA romidepsin (REDUC Part B Study) was able to show a modest (38%) reduction in VOA reservoir measurements (see Section 1.3); however, this did not correlate with a delay in rebound after treatment interruption (49, 50). This result indicates that a larger decrease in the reservoir is likely required to allow for continued suppression of virus off therapy. Another vaccine clinical study using Tat protein as the immunogen in chronically suppressed individuals showed approximately 90% decrease in proviral DNA reservoir and improved immunological profile (CD4+/CD8+ T-cell ratio) (194). Alternative viral reservoir measurements or treatment interruption were not performed in this study; however, this work indicates that the immune system in chronically suppressed individuals can be modulated by a therapeutic vaccine.
Unlike the Vacc-4x or Tat studies, a recent study in rhesus macaques using an adenovirus vector expressing SIV gag-pol-env provided in combination with TLR-7 stimulation was able to demonstrate effects after rebound (119). In this study, which induced robust Gag, Pol, and Env specific CD8+ and CD4+ responses, a slight delay in viral rebound and a decrease in viral setpoint was seen after treatment interruption. Clinical studies looking at adenoviral vectors and TLR-7 stimulation as curative therapy are still underway.
3.6 Gene Therapy
Gene therapy strategies that engineer functional changes into HIV or host genes have also been pursued and the ability of these strategies to effectively decrease the existing viral reservoir in the tissues of HIV-infected individuals remains to be determined. Because CCR5 is expressed on host CD4 cells and is a necessary recognition element for HIV infection, several of these efforts have focused on CCR5. Among them, Zn finger nucleases (ZFNs) are engineered nucleases that target and cut specific cellular DNA sequences. These breaks are then repaired by error prone pathways which can ultimately result in loss of function for the gene in question. ZFNs have been developed both for the editing of CCR5 alone (195, 196) and for dual CCR5/CXCR4 editing (197, 198).
A more flexible approach with respect to ZFNs is CRISPR/Cas9. Cas9, an endonuclease, performs the sequence-specific DNA double strand break, which is directed by a guide RNA (gRNA). The provirus in HIV+ cells, rather than ubiquitous cellular receptors like CCR5/CXCR4, can then be targeted with this technology. One strategy targets the HIV-1 long terminal repeat at the two end of the viral genome, with the goal of excising the proviral HIV-1 genome in its entirety (199-201). While this expected outcome has been observed in some studies (202); the opposite results were also reported resulting in virus escape (203-206). Other approaches target the provirus via short insertion and deletions (indels) caused by the error-prone nonhomologous end joining (NHEJ) pathway (203, 207-209), excising several different sites throughout the HIV-1 genome, individually or simultaneously (210). While this approach has worked in vitro to stop replication of HIV-1, the main challenge remains the delivery of therapy to target cells within an infected individual.
4 Conclusions and Outlook
HIV eradication research spans a broad scope of modalities, molecular mechanisms, and strategies. The field is at an exciting stage as tool compounds developed for other indications have enabled experimentation in vitro, in animal model systems, and in the clinic. As promising mechanisms for flush and kill emerge from these studies, it is likely that existing drugs will be repurposed for this use. In addition, opportunities will almost certainly arise for new medicinal chemistry and biologics campaigns directed at the optimization of agents specifically for HIV eradication.

