
Medicinal Chemical Biology
Abstract
Arguably, the two most important decisions facing drug discovery teams are selecting the target and the therapeutic modality as the focus of their research efforts. This article highlights the recent technical advances in medicinal chemical biology that strongly influence these early, yet centrally important, choices. The article is not meant to be a comprehensive review of all innovations in the area. Examples are chosen that illustrate the impact chemical biology has had recently on drug discovery research. Medicinal chemical biology is ideally suited to expand druggable space through the development of target identification and validation technologies. Equally, innovative therapeutic modalities are required to enhance the medicinal toolkit to ensure that high-quality clinical candidates are delivered that effectively target pathogenesis. Opportunities and challenges are described that serve to inspire further therapeutic research at the chemistry–biology interface.
1 Introduction
Drug discovery relies on the successful integration of multiple scientific disciplines across the physical and life sciences (Figure 1a). Medicinal chemistry is an intrinsically multidisciplinary endeavor that harnesses cutting edge molecular design, computational techniques, and synthetic chemistry to craft pharmacological agents that balance desirable pharmacokinetic and pharmacodynamic qualities to enable fundamental biological questions and therapeutic hypotheses to be addressed. Indeed, the interrogation of biological complexity necessitates multi- and cross-disciplinary methodologies. Chemical biology is increasingly influencing drug discovery and development, and its broad impact on biomedical R&D is growing. Likewise, chemical biology benefits from fundamental advances in the chemical sciences, and medicinal chemistry in particular. Chemical biology–medicinal chemistry interconnectedness has naturally evolved into a discipline I term medicinal chemical biology that describes the creation and application of chemical methods to not only interrogate biology and pharmacology but also to advance innovative therapeutic modalities.

Figure 1b highlights the existing and future opportunities for medicinal chemical biology in a traditional drug discovery paradigm. From target selection to clinical biomarker development, chemical biology has the potential to deeply influence therapeutic discovery. Chemical biology is also ideally suited to the advancement of biotherapeutics and vaccines because many technological advances, such as rational structure-based molecular design and selective biomolecule labeling chemistries, can be applied to their creation and optimization. This article describes recent innovations in medicinal chemical biology that are revolutionizing the discovery and development of transformational medicines.
2 Drug-target Engagement and Chemoproteomics
The main reason for drug attrition in clinical trials is a lack of efficacy in Phase II (1). Often, this results from a lack of target engagement by the drug, or modulation of the chosen target does not lead to the desired efficacy (or a combination of both factors). To advance a candidate drug through clinical trials, high confidence in rationale is ideally established early because the cost of failure at later stages of development is immense. Therefore, target identification and proof of target engagement must be established in order to build a high confidence therapeutic program. Although it should be appreciated that target identification and engagement are not absolutely essential for drug approval, it is less likely that a clinical candidate will be a successful drug without them.
Target identification and validation often rely on the application of chemical biology techniques. For example, the therapeutically relevant targets of compounds that result from phenotypic screens need to be deciphered to establish target-based discovery paradigms (2, 3). Such compounds are often derivatized to enable target isolation followed by mass spectrometry (MS)-based protein identification (more details below). Table 1 summarizes the recent developments in chemical probe and label-free target engagement technologies, which are further described in the following sections.
| Technology | Advantages | Challenges/disadvantages |
|---|---|---|
| Resin affinity enrichment | Various linkage chemistries available; simple protocol; many probes can be immobilized to enrich different proteins in one experiment; prior target knowledge not necessary | Only lysate; SAR development required to optimize resin-linkage chemistries; difficult to isolate membrane proteins; difficult to configure into a high-throughput screen |
| Chemical probe labeling | Direct assessment of engagement in live cells (often using click chemistry); all engaged proteins are enriched and analyzed (incl. Membrane proteins); high-throughput assay feasible; appending a dye enables in vivo imaging | SAR optimization required; competition labeling of covalent probe by non-covalent drug dependent on kinetics (faster offset compounds may appear less potent) |
| Quantitative energy transfer | Measures binding kinetics; amenable to high-throughput screening | Often requires an engineered cell line (known target proteins) and development of a functional probe |
| Cellular thermal shift assay (CETSA) | Uses parent drug; high-throughput screening format enables small molecule screening | Indirect. Thermal shift not always observed upon ligand binding (false negatives) |
| Drug affinity responsive target stability (DARTS) | Uses parent drug (label free); amenable for use with MS readout for unbiased target (and off-target) identification | Indirect assessment of engagement; ligand binding may not perturb proteolysis; suitable only in lysate |
Similar chemoproteomic technologies have been used to build pharmacokinetic–pharmacodynamic (PK-PD) relationships. Quantification of drug-target engagement is necessary to understand the levels of binding site occupancy that are required to drive a functional effect since these values are ultimately used to estimate the clinically efficacious dose. Additionally, clinical occupancy biomarkers are used to provide confidence that the target was engaged by the compound under investigation. A translational pharmacology framework was developed previously inspired by attrition data from clinical trials (4) that we termed the 4 pillars (Figure 2) (5). The guidance strongly advocates for a thorough understanding of target engagement for effective target validation. In fact, the lack of such information has led to the incorrect use of chemical probes in target validation experiments (6). Confirmation of target engagement at the site of action and quantification of target occupancy (Pillar 2) has been made accessible through advances in chemical biology, and several examples are provided below.

2.1 Affinity-based Methods for Target Engagement
Small molecules chemically attached to resins such as agarose beads enable binding proteins to be affinity enriched from complex proteomes (Figure 3) (7). Alternatively, a biotinylated small molecule of interest is exposed to a proteome in solution and subsequently immobilized on an avidin-coated bead. The advantage of this approach is that the biotinylated compound may be used directly in cell-based assays to ensure that the reporter attachment point does not impact the observable phenotype and thus perturb target isolation. Bound proteins are washed off the beads and identified/validated by MS or Western blot. On-bead digestion using trypsin enables peptide fragments to be analyzed using MS proteomics, and the original binding proteins are elucidated by the unique peptide sequence (8). Nonspecific interactions exist between proteins and the resin, and binding specificity may be confirmed by preincubating the proteome with the parent small molecule as it competes isolation of the proteins by the functionalized beads. A useful negative control for target identification/validation experiments is a structurally related, physicochemically matched, inactive compound that does not compete enrichment of the candidate binding proteins in a dose-related manner (5).

A classic example of successful target identification using affinity purification was that of thalidomide, an infamous antiemetic drug that possesses teratogenic effects. A thalidomide derivative was attached to beads to enable affinity isolation and analysis of binding proteins from cell lysate (Figure 4) (9). Cereblon, an E3 ubiquitin ligase, was identified and validated as the relevant target of thalidomide. Although the story of thalidomide is a desperately sad one, elucidation of its mode of action using chemical biology has advanced a novel therapeutic modality. Small molecule ligands conjugated to thalidomide colocate binding proteins with cereblon, resulting in ubiquitination and proteasomal degradation of the target (see Section 3.1 for more details on these proteolysis-targeting chimeras, or PROTACs) (10, 11).

The following recent example serves to illustrate a traditional pull-down target identification paradigm that has yielded a new anticancer therapeutic target. A phenotypic screen was developed to identify inhibitors of the HSF1 pathway, a master regulator of the heat shock response involved in tumorigenesis (12, 13). A bisamide hit was optimized to CCT251236 (Figure 4) that possessed desirable PK-PD to establish efficacy in a human ovarian carcinoma xenograft model (12). Profiling in kinase biochemical screens and the CEREP diversity panel did not clearly identify targets that could explain the efficacy of CCT251236 and therefore an unbiased chemoproteomic strategy was pursued. A vector in the bisamide series was identified in the SAR study that suggested it could serve as the attachment point to the resin to enable a pull-down experiment from cell lysate. Quantitative MS proteomics using SILAC yielded three candidate binding proteins, and the putative transcription factor regulator and iron-binding protein pirin was the only validated hit using competition binding experiments (with active and inactive control molecules) and biophysical methods, including surface plasmon resonance (SPR) using recombinant protein. A cocrystal structure revealed an interesting interaction between the bisamide functionality in CCT251236 and a water molecule that coordinates the metal ion bound in pirin. Although the role of pirin needs to be further established, CCT251236 phenocopied the inhibition of tumor cell migration observed previously using a different pirin ligand. As a result of this work, the chemical probe toolkit to investigate pirin function has been expanded: see also http://www.chemicalprobes.org/cct251236 and Section 3.1 on PROTAC. Many other successful examples of target identification using affinity-based pull-downs have been reported and are reviewed elsewhere (7, 14-16).
Affinity-based methods are also useful in the development of drug-target occupancy biomarkers. For example, a resin-bound purvalanol B reagent was used in our group to isolate the kinase TrkA, an important cancer and pain target, from cell lysate (Figure 4) (17). Quantification of TrkA levels was enabled using stable isotopically labeled TrkA peptide fragments in a novel multiple-reaction monitoring MS protocol. Isolation of TrkA was competed in a dose-dependent manner by kinase inhibitors illustrating the utility of the technology for measuring TrkA engagement. Additionally, purvalanol B binds many other kinases (18), and so the approach could be expanded to accurately measure occupancy of other kinases by drugs and drug candidates. A matrix created by the immobilization of multiple promiscuous kinase inhibitors, including purvalanol B, has enabled the development of a quantitative chemoproteomic technology to assess kinome selectivity in cell lysate (19).
Another affinity-based approach employs a quantitative energy transfer assay that utilizes a cell-permeable functional chemical probe containing a BODIPY motif and a 19-kDa-luciferase (NanoLuc)-tagged donor target protein (20). Real-time bioluminescence resonance energy transfer (BRET) between the fluorescent tag and the NanoLuc protein is then competed in dose by the compound of interest to provide an assessment of live cell target occupancy and binding kinetics (Table 1). The approach has been used successfully to determine histone deacetylase activity of a series of inhibitors in live cells, which helped explain the prolonged duration of the action of the prodrug FK228 (20). The NanoBRET technology was used recently to measure the selectivity of clinical kinase inhibitors against 179 full-length kinases (21). Through this work, the in-cell selectivity of crizotinib (approved for the treatment of some non-small cell lung cancers) was found to be better than assessments of selectivity using traditional biochemical assays due to high intracellular ATP concentrations that hinder drug-target engagement of certain kinases. Related techniques have been developed recently that avoid the need for an engineered protein using a high-quality antibody for the target (TR-FRET) (22), or through metabolic labeling of cell surface receptors (GlycoFRET) (23).
Due to the presence of posttranslational modifications (PTMs), protein–protein interactions (PPIs), subcellular compartmentalization, and high-affinity competitive metabolites, drug-target occupancy in live cells could be significantly different compared to cell lysate. Affinity resins are not cell permeable of course which is a deficiency of the technique (Table 1). Additionally, biotinylation and fluorescent tagging of chemical probes substantially alter their physicochemistry (such as molecular weight, hydrogen-bonding capacity, and lipophilicity) that may affect permeability and subcellular distribution. A small “click” handle may be incorporated as a “silent” reporter into the chemical probe structure to enable cellular permeation and subsequent assessment of engagement in a more physiologically relevant context of an intact cell. Click chemistry has revolutionized chemical biology due to the ease with which molecular transformations can be made in biological, and aqueous, systems (24). For instance, a copper(I)-mediated azide–alkyne cycloaddition (CuAAC) reaction enables addition of a dye or biotin or FLAG tag in cell lysate following protein engagement by the probe in intact cells (Figure 5) (25-27). This synthetic methodology has been used successfully to enable drug-target engagement experiments in cells. NS5A was identified as the putative target of a series of compounds with antihepatitis C virus (HCV) activity using genetic profiling of resistant mutants that emerged following prolonged incubation of the lead compound with replicon cells (28, 29). A biotinylated derivative was used to affinity isolate NS5A, and streptavidin-mediated enrichment was competed by the presence of the parent compound to confirm the specificity of target engagement. This work led to the development of the drug daclatasvir, an NS5A inhibitor for the treatment of HCV (30). To further validate NS5A as the relevant target, studies were initiated in our group using functional chemical probes for cell imaging experiments (31). Interestingly, BODIPY-conjugated fluorescent NS5A inhibitors that retained anti-HCV activity (albeit with less potency) did not appear to colocate with the protein using immunohistochemistry likely due to the high molecular weight and lipophilicity that perturb subcellular distribution of the probe (Figure 5). However, a more potent inhibitor incorporating a silent azide click reporter was incubated with cells, which were then fixed/permeabilized and an alkyne-tagged dye subsequently appended to the probe using CuAAC that showed complete colocalization with the NS5A protein. We developed further mechanistic cell-based imaging techniques to reveal that the NS5A inhibitors possess an unexpected phenotype by shuttling the target to nonfunctional lipid droplets in replicon cells (32). Target validation using clickable imaging-based chemical probes in this way has become an important technique that has become widely used in chemical biology (33).

Since the development of CuAAC, many other examples of biorthogonal click chemistry reactions have emerged (34). Tetrazine click ligations have set new standards of efficiency with extremely high rates of reaction resulting in its widespread use (35). These reactions rely on an inverse-electron demand Diels–Alder reaction between a 1,2,4,5-tetrazine and a strained alkene (Figure 6). In one recent example of the approach from our group, structure-based design of a trans-cyclooctene (TCO) probe based on the potent HDAC inhibitor largazole enabled measurement of HDAC1 and 2 target engagement by a different inhibitor called dacinostat (38). Cells were treated with TCO-largazole, lysed and exposed to tetrazine-biotin to enable HDAC isolation and analysis. Competition using different doses of dacinostat enabled quantification of in-cell target occupancy. Interestingly, HDAC3 was not isolated from cells, even though the TCO-largazole probe was found to be a potent inhibitor in a biochemical assay, suggesting that HDAC3 occupancy is context dependent.

2.2 Target Engagement Using Reactive Chemical Probes
Affinity-based methods of measuring drug-target engagement either use cell lysate or rely on very potent functional probes that can be used in intact cells for the reasons described above. Reactive functional probes are ideally suited to live cell, and thus more physiologically relevant, applications because covalent protein adducts are more resilient to cell lysis. This approach is particularly important for integral membrane proteins because cell lysis may destroy the native conformations of the target that are required for binding interactions.
Activity-based protein profiling (ABPP) utilizes a related covalent strategy (39). A functional activity-based probe (ABP) incorporates a protein-binding element for the target or target family of interest, a reporter (a fluorescent dye, biotin, or click handle) and a reactive warhead that is selected to specifically engage an amino acid residue that is involved in the activity of the protein. Often, the ABP is designed to be promiscuous so that the activities of many related proteins are measured using MS proteomics.
ABPP is context dependent because changes in the activity of the protein, say from PTM of a catalytic residue, perturb labeling by the probe (40). ABPP has been used successfully to profile drug-target engagement across several gene families and opportunities to use the technology for clinical biomarker development are being explored (41).
The following sections describe the use of different reactive warhead chemistries in the design and development of activity (and reactivity)-based protein profiling technologies that are of relevance to drug discovery research (42). Some examples were originally developed as proof-of-concept studies using lysate, but then future innovations led to the creation of probes that were suitable for intact cell labeling.
2.2.1 Cysteine Targeting
The thiol motif present in cysteine makes this a highly nucleophilic amino acid, and many covalent small molecules have been developed to target the residue using a variety of electrophiles (Figure 7) to enhance pharmacological duration (43). Additionally, the site-specific targeting of one cysteine over another has led to the development of selective agents (44). An important advantage of targeting cysteine is that mutation to the physicochemically similar serine often retains the original function of the protein, but the covalent probe loses its activity due to a significant reduction in nucleophilicity of the residue. This is harnessed in target validation experiments since the cell line possessing the mutation should then be resilient to phenotypic perturbation in the presence of the covalent inhibitor (43).

There are notable examples of such probes in the kinase field, and these have led to the development of successful clinical agents (43). The acrylamide in the anticancer drug ibrutinib targets the reactive Cys481 residue in the ATP site of BTK (45, 46). A fluorescent covalent chemical probe based on the ibrutinib structure was used to measure target engagement clinically as a pharmacological (Pillar 2) biomarker that provided mechanistic confidence for the drug development program (Figure 8) (47). A similar approach was used recently to build PK-PD and selectivity understanding for a different covalent BTK inhibitor (48) and inhibitors of EGFR (49) and JAK3 (50). Using an alternative technology, a fluorescent covalent inhibitor of BTK was employed as a companion imaging probe (CIP) with fluorescence polarization microscopy to measure target occupancy (51). In another study, a selective RSK inhibitor (FMK) was designed to engage Cys436 using a fluoromethyl ketone warhead, and a clickable probe (FMK-PA) assessed intact cell target engagement (Figure 8) (52). Inspired by the BTK, RSK, and related studies, we mapped the cysteine residues in the kinase ATP site to inventory a “kinase cysteinome” using structural alignment and bioinformatics analyses (43). This work identified 18 spatial positions in 200 kinases, each one thus serving as a potentially targetable site for future development of covalent small molecule drugs and companion target occupancy probes.

A clickable version of ibrutinib was used to assess off-targets in an unbiased manner using quantitative MS proteomics as a readout, and a number of non-kinase targets were identified (53). Related approaches have been used to identify targets of cysteine-reactive natural products such as sulforaphane and andrographolide through the incorporation of click reporter functionalities (54, 55). In a study of the protein-wide cysteinome, hyperreactive cysteine residues were identified using a promiscuous clickable iodoacetamide probe in conjunction with quantitative MS proteomics (56). These studies identified many targetable cysteines in proteins often termed “undruggable,” thus broadening the opportunities for pharmacological modulator development. Fragment-based drug discovery often utilizes biophysical screening of weak-binding fragments at high concentrations such that chemical space is efficiently covered in a small library of compounds. A cysteine-reactive fragment library was screened in live cells using the abovementioned chemoproteomic assay since cysteine engagement by the fragment prevented labeling of that site by the clickable iodoacetamide probe (57). More than 700 cysteines were labeled by reactive fragments, many of which do not possess small molecule probes. The protein hits were distributed across druggable gene families and other targets deemed undruggable such as transcription factors and scaffolding complexes.
Intrinsically reactive protein cysteines undergo facile oxidation, particularly under conditions of oxidative stress such as those that exist in inflammatory diseases. Chemical probes have been developed to specifically target these species. Dimedone carbon nucleophiles react with the electrophilic sulfenic acid moiety (58), while aryl nitroso compounds were designed to target sulfinic acid residues (59). Cysteine redox chemistry is particularly important for cysteine-targeting probes and drugs since oxidation of the residue may prevent target engagement, as suggested for the EGFR inhibitor afatinib (60). Probes bearing dimedone warheads, such as DYn-2, enabled the oxidized cysteinome to be mapped and understood using chemoproteomics that allowed differences between redox states to be assessed (Figure 9) (61, 62). A fragment-based approach similar to that described above but using a library of carbon-based nucleophiles enabled the ligandable sulfenome to be elucidated, thus highlighting targetable residues that are dependent on protein redox states (63). For instance, pyrrolidinediones were found to engage protein tyrosine phosphatases, enzymes that have been traditionally difficult to drug. Of particular relevance to drug metabolism, the heme thiolates of P450s were recently found to be redox regulated via sulfenic acid formation (64).

2.2.2 Beyond Cysteine Targeting
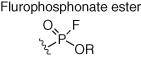
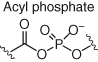
Although cysteine-targeted probes have found considerable utility in chemical biology and medicinal chemistry, cysteine residues are underrepresented in protein functional sites. Additionally, due to the intrinsically high reactivity of cysteine, this complicates the development of site-specific modulators. As a result, probes are required that engage residues beyond cysteine (Table 2). Serine hydrolases possess a catalytic serine residue that is particularly reactive due to lowering of the hydroxyl pKa in the microenvironment of the binding site. Fluorophosphonate (FP) esters react with the nucleophilic serine, and promiscuous ABPs utilize this warhead (and a reporter such as biotin or rhodamine dye) to canvass serine hydrolase occupancy in complex proteomes (Figure 10) (65). The selectivities of numerous serine hydrolase inhibitors, bearing a variety of electrophiles, have been profiled using ABPP (78, 79). Similarly, kinase ATP-site occupancy was measured using an acyl phosphate desthiobiotinylated probe that was designed to react with the conserved lysine residue (Table 2, Figure 10) (67). When used with competitive MS proteomics, the probe was able to report on kinase inhibitor selectivity in proteomes. This robust technology is widely used in the pharmaceutical industry to advance the design of selective kinase inhibitors (80). The technique was also used to identify a tumor-specific point mutation of the catalytic aspartic acid in the active site of CSNK1A1 (81). Although this chemoproteomic approach delivers a more physiologically relevant assessment of kinase inhibitor selectivity (compared to biochemical assays), the impermeability of the probe restricts its use to cell lysate. It is preferable to measure in-cell target engagement and selectivity and therefore cell-permeable probes are required.
| Reactive group | Targeted amino acids | Examples of targeted proteins |
|---|---|---|
 |
Serine, tyrosine | Serine hydrolases (65), glutathione S-transferase (66) |
 |
Lysine | Kinase conserved lysine (67) |
 |
Lysine | Kinase conserved lysine (68, 69) |
 |
Lysine, tyrosine, histidine, serine threonine | Serine hydrolases, DcpS, transthyretin, GPCRs, P-glycoprotein, glutamate dehydrogenase, ATPase (70) |
 |
Lysine, tyrosine, serine | DcpS, transthyretin, intracellular lipid-binding proteins (71) |
 |
Glutamate | PDE6δ (72) |
 |
Lysine | Non-catalytic lysine in CDK2 kinase (73) |
 |
Lysine, tyrosine | Glutathione S-transferase, NME1/2 kinase, glutamate dehydrogenase (74, 75) |
 |
Histidine | Isocitrate dehydrogenase 1 (76) |
 |
Methionine | Calmodulin (77) |

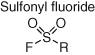
Sulfonyl fluoride chemical probes have found widespread utility in chemical biology research (70, 82). The electrophile possesses suitable aqueous stability for use in biological milieu and context-dependent reactivity with several amino acid residues beyond cysteine (tyrosine, lysine, serine, threonine, and histidine) in functional protein sites (Table 2). This is due to activation of the warhead via hydrogen bonding to the fluorine atom that occurs in the microenvironment of the binding site. Simple molecules containing a sulfonyl fluoride were originally identified as covalent inhibitors of serine hydrolases (83), and then the warhead was incorporated into several reversible protein binders, thus converting them into irreversible inhibitors (84-86). A sulfonyl fluoride-containing ATP derivative called FSBA (fluorosulfonyl benzoyl adenosine) was designed to map nucleophilic residues in ATP-binding proteins (Figure 11) (87) and subsequently developed as a promiscuous kinase inhibitor due to its reactivity with the conserved lysine residue in the ATP site (88). FSBA served as inspiration for the design and development of a cell-permeable reactivity-based probe called XO44 for determining kinase inhibitor selectivity in live cells (Figure 11) (89). XO44 retains the lysine-targeting sulfonyl fluoride warhead incorporated into a promiscuous hinge-binding aminopyrazole scaffold and a terminal alkyne click reporter that enables kinase enrichment and MS proteomic analysis. XO44 isolated 133 kinases in the Jurkat cell line. The chemoproteomic technology was used to show for the first time that the anticancer kinase inhibitor dasatinib possesses selectivity for SRC over LCK in intact cells (selectivity determination using cell lysate-based methods suggested that the drug was not selective). XO44 also identified 420 non-kinases, suggesting that these proteins are targetable using kinase-like motifs. XO44, when used in conjunction with MS proteomics, may thus enable selectivity determination of compounds across a considerable fraction of the ATP-binding proteome.

A sulfonyl fluoride occupancy probe (SF-p1-yne, Figure 11) was developed to engage a tyrosine in the binding site of the mRNA decapping scavenger (DcpS) enzyme that lacks a cysteine residue (90). This was the first example of rationally targeting a specific tyrosine amino acid using a chemical probe. SF-p1-yne was used to validate DcpS as the relevant target of a series of diaminoquinazoline (DAQ) derivatives that were in development for the treatment of spinal muscular atrophy. The physicochemistry of some compounds (lipophilic and dibasic) resulted in lysosomal accumulation (91) that partially hindered engagement of DcpS, as shown in cell-based target occupancy experiments using SF-p1-yne. A sulfonyl fluoride covalent modulator of transthyretin (TTR, a protein involved in amyloid diseases) was designed to react with a pKa-perturbed lysine in the binding site and stabilized the protein to prevent fibril formation (92). Sulfonamide formation resulted in concomitant turn-on fluorescence that was used to measure the kinetics of adduct formation with the protein.
Sulfonyl fluorides are beginning to find utility as reagents to stabilize difficult-to-crystallize proteins that aid structure-based drug design. A sulfonyl fluoride covalent inhibitor of the adenosine A1 GPCR stabilized the protein by 16 °C that facilitated cocrystallization studies (93). The binding site of a covalent sulfonyl fluoride peptidic inhibitor of IL17 was mapped using peptide MS that led to successful structure-enabled design of a macrocyclic inhibitor with impressive cell-based activity and metabolic stability (94). The sulfonyl fluoride motif will likely be incorporated into phenotypic screening hits to facilitate target identification efforts in the future, particularly as the fragment does not significantly increase the lipophilicity of the compound (ΔLogP 0.17), thus limiting nonspecific binding (90).
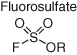
Fluorosulfate electrophiles (Table 2) possess ameliorated reactivity relative to the sulfonyl fluoride motif, due to resonance stabilization from the additional oxygen atom, but with similar chemoselectivity (generally preferring tyrosine and lysine) (71). A fluorosulfate TTR modulator, as for the sulfonyl fluoride derivative described above, formed a fluorescent adduct with the protein through reaction with the binding site lysine, which was used to assess target engagement in vivo (95). A fluorosulfate-containing DAQ derivative FS-p1 (Figure 11) was found to react with DcpS, as for the sulfonyl fluoride derivative, but the probe unexpectedly engaged a noncatalytic serine in the binding site (96) (the only other known example of covalent targeting of a noncatalytic serine is aspirin acetylation of cyclooxygenase enzymes) (97). FS-p1 also demonstrated considerably improved chemical and metabolic stability over the sulfonyl fluoride congener, suggesting that the fluorosulfate warhead is suitable for covalent inhibitor drug design. The improved stability was also harnessed recently in the development of an unnatural amino acid-based technology to map PPIs. Fluorosulfate-l-tyrosine was genetically encoded into proteins that enabled proximity-induced inter- and intraprotein cross-linking with tyrosine, lysine, and histidine residues (98). The approach has the potential to canvass changes in protein networks that are remodeled in disease.
Fluorosulfate probes were developed to exemplify a strategy termed “inverse drug discovery” (99). Clickable drug-like compounds that incorporated fluorosulfate warheads were exposed to cells, and adducted targets were affinity enriched and identified using MS proteomics. New chemical probes of HSDL2 (an emerging glioma target) and the lipid-binding protein CRABP2 were discovered (100). Profiling of a larger library of reactive probes would further demonstrate the utility of the strategy and potentially expand druggable space.
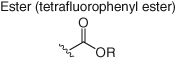
Broader profiling using a promiscuous amine-reactive tetrafluorophenyl ester clickable probe, in a manner similar to the cysteinome approaches described above, mapped >9000 hyperreactive lysines across the human proteome (Table 2) (68). A small library of amine-reactive fragments was then screened using competitive MS proteomics to yield 121 ligandable lysine sites in 113 proteins. Hits included small molecules that allosterically inhibit enzymes and PPI modulators, thus illustrating the potential of the technology.
There are other emerging opportunities of targeting lysine residues in drug discovery and chemical probe development (101). For example, the first selective irreversible inhibitor of the lipid kinase and cancer target PI3Kδ was discovered recently employing an unusual strategy – covalent engagement of the conserved ATP-site lysine (69). Although the approach appears counterintuitive, the amelioration of lysine reactivity using a carefully designed ester warhead and optimization of equilibrium binding interactions with the protein yielded a long duration and highly selective inhibitor, as determined using chemoproteomics in live cells. The approach is noteworthy as it challenges the dogma that targeting conserved residues in proteins intrinsically leads to low specificity. Engaging residues beyond cysteine is particularly important in cancer due to the emergence of cysteine-to-serine resistance mutations in the clinic, as observed for covalent inhibitors of BTK and EGFR (102, 103).
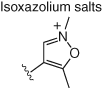
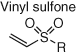
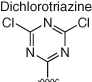
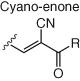
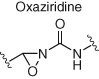
Several other electrophiles (Table 2) have been developed to target residues beyond cysteine such as glutamate (isoxazolium salts) (72), lysine (vinyl sulfone) (73), tyrosine/lysine (dichlorotriazines) (74, 75), histidine (cyano enone) (76), and methionine (oxaziridine, epoxide) (77, 104). The expansion of the toolbox for site-specific amino acid engagement using chemical probes and drug-like compounds will enable future chemical biology studies and unearth new opportunities for drug discovery (105). Advances in protein labeling chemistry will also enable the field of bioconjugation (Section 3).
2.2.3 Photoaffinity Labeling
Photoaffinity labeling relies on the use of high-energy ultraviolet light to reveal a highly reactive, and promiscuous, warhead that forms adducts with a plethora of bioorganic functionalities in proximity to the probe. Commonly used photoactive groups, the utilization of which has been reviewed elsewhere, include benzophenone, diazirine, and azide functionalities that form a CO diradical, carbene, or nitrene species, respectively, upon UV irradiation (106, 107). Photoaffinity labeling is ideally suited to unbiased target identification since prior knowledge of binding site is not required due to the highly reactive nature of the photoprobe. Some classic examples of successful photoprobe-based target ID/validation include the cotransins (Sec61α) (108), itraconazole (NPC1 and VDAC1) (109), pladienolide (SF3B) (110), and taxol (β-tubulin) (Figure 12) (111). Photoaffinity labeling of target proteins and competition by parent drugs in a dose-responsive manner may enable the development of target occupancy biomarkers, even for low-expressing membrane proteins (112). Photoaffinity labeling may suffer from low yields, and binding affinity may not correlate with protein labeling efficiency that can hinder the design and development of an effective probe. For this reason, we developed library chemistry and parallel screening of protein labeling to expedite the rapid selection of the optimal photoaffinity probe for DcpS (113).

Photoaffinity labeling not only provides opportunities to identify the target proteins of small molecules of interest, but it potentially also illuminates their binding sites. In one recent example, nonsteroidal anti-inflammatory drugs (NSAIDs) were armed with a diazirine photoaffinity warhead and terminal alkyne click reporter to label and enrich interacting proteins in live cells (114). Through the use of isotope recoding of the conjugated peptides and targeted MS-based assignment, hundreds of binding sites were characterized, some of which were confirmed using the cellular thermal shift assay (CETSA) (Section 2.3.1).
A library of fully functionalized fragment (FFF) probes that contain a protein-binding motif, an alkyl diazirine photoreactive tag, and a biorthogonal click reporter were exposed to intact cells, UV–irradiated, and cross-linked proteins identified using quantitative chemoproteomics (115). A fragment–protein interaction map was created that highlighted thousands of small molecule-binding site interactions in human cells. Structure–activity relationships were elucidated that confirmed the specificity of the fragment–protein interactions. For instance, a fragment hit was identified for the solute carrier membrane protein SLC25A20, which led to the development of an elaborated micromolar inhibitor of the transporter in cells. Proteome-wide fragment-based drug discovery will likely find considerable utility as a hit generation/target identification technology in the future, particularly as larger libraries are profiled in a wider variety of cell lines.
2.3 Target Engagement Using Label-free Methods
Chemical probe-based protein labeling has delivered useful target engagement and hit generation technologies. However, SAR generation is required to ensure that the designed functional probes retain target protein-binding capabilities. Therefore, orthogonal label-free methods potentially accelerate target identification and validation studies (Table 1) (116).
2.3.1 Cellular Thermal Shift Assay
Small molecule-target engagement often leads to a shift in the melting temperature of the binding protein due to stabilization, or destabilization, upon ligand binding. High-throughput in vitro thermal shift assays use a dye that fluoresces upon binding nonspecifically to hydrophobic regions of the protein that are exposed during thermal denaturation. Recently, a CETSA was developed using intact cells and lysate (117). A whole proteome is heated in the presence of the small molecule, aggregated proteins are removed by centrifugation, and the thermal stability of remaining proteins in solution is assessed by WB or MS techniques. To validate the sulfonyl fluoride probe-based target occupancy measurement of the DcpS inhibitors in human primary cells described in Section 2.2.2, CETSA was used as an orthogonal, confirmatory method (118). The enzyme affinity of 10 nM for the lead DAQ inhibitor using CETSA was nearly identical to that found using the chemical probe approach (11 nM).
2.3.2 Drug Affinity-responsive Target Stability
Drug affinity-responsive target stability (DARTS) relies on protein ligands being able to reduce target proteolysis in cell lysate (119). A recent example explored the oncogenic Wnt/β-catenin inhibitory mechanism of action of the multireceptor tyrosine kinase inhibitor axitinib (120). Treatment of cells with axitinib stabilized several proteins from pronase proteolysis, which were resolved using 2D gel electrophoresis and analyzed via MS. The E3 ubiquitin ligase SHPRH was found to be the functionally relevant target of axitinib for negative regulation of Wnt/β-catenin signaling, independent of the drug's kinase activity. SHPRH was then confirmed as an axitinib target using CETSA with WB analysis. DARTS has been used successfully to assess small molecule–protein interactions for methotrexate–DHFR (121), olaparib–PARP (121), rapamycin–FKBP12 (119), betulinic acid–GRP78 (122), piperlongumine derivative CG-06-STAT3 (123), and an inhibitor of the glutamine transporter ASCT2 (124). The technique is also applicable to the identification of metabolite-binding proteins. α-Ketoglutarate, which extends lifetime in Caenorhabditis elegans by dietary restriction, was shown to bind to, and inhibit, ATP synthase subunit β using DARTS (125).
3 Emerging Therapeutic Modalities
Target identification and validation using the technologies described above enable the medicinal chemical biologist to directly address one of the major challenges facing the drug discovery industry – therapeutic target selection. An area that is equally important but has received much less attention is that of therapeutic modality selection. The following section is not meant to exhaustively describe all therapeutic modalities influenced by innovations in chemical biology. Rather, vignettes have been selected that exemplify the emerging opportunities for medicinal chemical biology. From large molecules such as vaccines and antibodies to emerging small molecule approaches such as targeted protein degraders and molecular glue, the various ways in which the medicinal chemical biologist can tackle disease is vast (see Figure 13 for some examples). With a larger palette of modalities to choose from, it is hoped that drug discovery teams will be able to select the best modality that delivers the desired therapeutic outcome.

3.1 Protein Degradation
PROTACs are bifunctional molecules that consist of a target protein-binding motif and a ligand for an E3 ligase, such as the cereblon ligand thalidomide described above (126). PROTACs colocate the target and the ligase machinery, resulting in ubiquitination and subsequent proteasomal degradation of the target. PROTACs thus mimic genetic methods of protein downregulation that deplete PPIs and noncanonical signaling events, but with improved delivery as expected for a small molecule. PROTACs may possess additional efficacy over traditional small molecule inhibitors of protein function, and the mode of action is in theory catalytic; once a PROTAC molecule has bound and degraded its target, it is then available to repeat its function (127). PROTACs are also able to degrade various resistance mutations in cancer, provided binding to the mutant form of the protein is retained (128). For these reasons, PROTACs have received considerable attention in the drug discovery arena recently, and several companies have been created to exploit and further develop the modality (129).
In one recent example, a PROTAC was developed based on the structure of CCT251236, previously described as an inhibitor of the HSF1 pathway and putative pirin inhibitor (see Section 2.1, Figure 4) (12). The cereblon-targeting thalidomide conjugate CCT367766 (Figure 14) was developed to confirm in-cell target engagement of pirin (130) since binding to the bisamide series was shown originally using resin-based pull-down chemoproteomics in cell lysate (Section 2.1). Pirin was the only target degraded in cells by CCT367766 as confirmed using MS proteomics, and inactive control compounds did not reduce protein levels. Equally, at higher concentrations, a hook effect was observed, as expected for the formation of a ternary complex, and CRISPR/Cas9 knockout of cereblon ablated the effect. This work nicely illustrates the potential utility of the PROTAC chemical methodology to validate phenotypic screening hits in live cells (particularly for understudied targets with no known biomarkers or catalytic activity) that complements other technologies described in Section 2.

The PROTAC field will need to broaden the scope of targeted proteins beyond those that have been intensively studied thus far (mostly bromodomains and kinases). Unbiased ligandability profiling using chemoproteomics yields interaction maps such as those described in Section 2.2.3 and can provide one means by which ligands for new degradation targets could be identified. Since ligandable E3 ligases are potentially unearthed simultaneously in the same cell type, it could provide confidence in the selection of target–ligase pairs for subsequent design of heterobifunctional PROTACs. Importantly, neutral binding ligands identified through such approaches can be converted into functional degraders through conjugation to E3 ligase ligands.
The rational design of PROTAC heterobifunctional conjugates opens new opportunities for medicinal chemistry to be applied to the control of target protein levels. Unfortunately, it is challenging to balance both on-target and ligase pharmacology in a single molecule while maintaining appropriate oral drug-type properties (molecular weights are considerably higher than monovalent drugs, for instance) (131). Interestingly, there are several emerging examples of small molecule ligands that trigger downregulation of their target proteins. Certain ATP-site kinase inhibitors appear to be able to deprive the client kinase from the Hsp90-Cdc37 chaperone system, resulting in ubiquitination and subsequent target degradation (131, 132). An inhibitor of PI3Kα called GDC-0077 (currently in Phase 1) was recently shown to promote degradation of the mutant enzyme in vivo over the wild-type form of its target and may drive a component of its efficacy by inhibiting pro-oncogenic PPIs (133). Another report from our group described the intriguing selective transcriptional downregulation of JAK2 and JAK3 in human immune cells by a pan-JAK inhibitor (134). These observations may be a result of phospho-STAT (the products of JAK enzyme activity) directly controlling JAK expression levels through binding of the transcription factor to the kinase promoter regions (135). It is possible that many kinases control the regulation of their own genes (136), and more research is required to explore the possible generality of these effects and their therapeutic potential. Conversely, protein ligands may cause stabilization of their binding targets. For example, FLT3 kinase site binders inhibit the canonical ATP-signaling function of the protein but increase membrane levels that may enhance pro-oncogenic scaffolding functions and the emergence of resistance (137).
Due to the ease with which protein levels can be measured using Western blot or MS, and the growing appreciation that many ligands cause target degradation or stabilization, then all clinical candidates and approved drugs should be screened for proteostatic or transcriptional effects (138). Although the rational molecular design of such features is currently not possible, deeper mechanistic insight that can be provided by chemical biology studies and the use of chemoproteomics to efficiently explore protein degradation propensity will no doubt enable future development of structure–degradation/stabilization relationships and influence target selection strategies. Additionally, drug discovery projects will often backup the candidate lead compound with a structurally differentiated derivative to address potential attrition from safety liabilities. Arguably, this strategy would realize greater return on investment if “backup” compounds differentiated on pharmacological mechanism (e.g. an inhibitor is followed by a “degrader”) since the major attrition in clinical trials is due to lack of efficacy in Phase II, and a degrader modality is more likely to phenocopy the efficacy seen in genetic KD/KO experiments as described.
3.2 Pharmacological Chaperones
Genetic disorders are caused by mutations that often result in a loss of function (LOF) of the protein product. LOF mutations can lead to a trafficking defect, where the nascent protein becomes destabilized, prematurely degraded and therefore less protein is appropriately localized in the desired cellular compartment (139). Pharmacological chaperones directly engage the misfolded protein and correct the trafficking defect (140). Cystic fibrosis (CF) is caused by LOF mutations in the CF transmembrane conductance regulator (CFTR), and small molecule chaperones increase the amount of protein at the cell surface. Lumacaftor (Figure 15) is one such drug that is used to treat CF, and it has been shown to directly engage CFTR by NMR, DARTS, and CETSA studies (141-143). An interesting, and paradoxical, phenomenon is that functional inhibitors may also act as pharmacological chaperones. Another drug, migalastat (Figure 15), is an inhibitor of α-galactosidase A (α-GalA), yet it also corrects the trafficking defect in the mutated enzyme that causes a lysosomal storage disorder called Fabry disease (144). PK-PD investigations are essential to understand the optimal dosing regimen for this modality (every other day in the case of migalastat) to ensure that the trafficked enzyme is not then continuously inhibited by the drug. Pharmacological chaperones are promising agents for the treatment of lysosomal storage diseases in particular because existing enzyme replacement therapy has limitations due to poor tissue distribution, particularly in the CNS (145). Other gene families are amenable to small molecule-mediated chaperoning, such as ion channels, serine hydrolases, solute carriers, and GPCRs. Mutations in MC4R cause severe early-onset obesity, and some inhibitors of the GPCR have been shown to also act as pharmacological chaperones (146). Interestingly, inhibitors, pharmacological chaperones, and agonists of MC4R possess remarkably similar structures and physicochemistry suggesting that subtle changes in target conformation induce a variety of functional effects (Figure 15). Broadly, monovalent proteostasis modulators (degraders and chaperones) possess interesting structure–function relationships that are not completely understood currently, and further chemical biology studies (such as deciphering the cellular chaperone (147)) are required to elucidate the molecular drivers of these attributes that could facilitate rational medicinal chemistry targeting. Additionally, pharmacological chaperoning of wild-type proteins may be a useful therapeutic modality but has received considerably less attention than conformational disorders (148).

3.3 Molecular Glue
Molecular glues are small molecule enhancers of PPIs (or interactions between domains of the same protein) that either serve to augment or inhibit signaling pathways. Several natural products discovered in phenotypic screens have been subsequently shown to possess a molecular glue mechanism. For instance, rapamycin is a bacterial macrolide with immunosuppressive and antiproliferative properties that binds the prolyl isomerase FKBP12 (149). The resulting complex creates a recognition interface for mTOR that hinders access of protein substrates to the kinase active site (150). A different immunosuppressant, the cyclic peptide and fungal metabolite cyclosporine, similarly binds a prolyl isomerase called cyclophilin, and the resulting complex inhibits calcineurin, a phosphatase involved in T-cell activation (151).
As described above, PROTACs induce the colocation of a target protein and an E3 ligase and, therefore, act as rationally designed molecular glues. Further work in this area will include the discovery and design of synthetic molecular glues with improved oral drug-type properties and reduced synthetic complexity than bifunctional conjugates or natural products.
Protein phosphatases have been extremely difficult targets for the pharmaceutical industry to crack due to the highly polar nature of the enzyme-binding site that needs to accommodate phosphorylated amino acid residues. An assay was developed recently to identify allosteric inhibitors of the important cancer target SHP2 phosphatase by screening for compounds that blocked activation by a phosphotyrosine peptide (152). The lead compound, SHP099, was subsequently found to glue different domains of the phosphatase together, preventing the free movement required for SHP2 enzymatic activity. Careful design of biochemical assays, and the further adoption of phenotypic screening, will likely yield additional small molecules that possess molecular glue mechanisms.
3.4 Small Molecule RNA Binders
Regulatory elements in mRNA called riboswitches bind small molecule metabolites that directly control protein expression. For instance, a riboswitch binds flavin mononucleotide (FMN) to control riboflavin biosynthesis, which has become a target for the development of new antibiotics (153). Interestingly, an antibacterial phenotypic screen fortuitously yielded ligands of the FMN riboswitch also (see ribocil, Figure 16) (154). These discoveries illustrate the potential druggability of RNA structures using small molecules. Indeed, recent advances using chemical biology have shown that microRNA and genetic disorders characterized by expansion repeats are targetable using small molecules (155, 156). The molecular design rules for targeting RNA continue to be developed by medicinal chemists, and due to the significant investments that have been made in this area in recent years, the huge untapped potential for the modality may be realized in the near future (157).

3.5 Synthetic Vaccines and Antibodies
Traditional vaccines are complex mixtures of attenuated bacteria and viruses, prepared using heat or chemical inactivation of the pathogen. Such methods often suffer from batch-to-batch variability and unproductive reactions to heterogeneous vaccine components. Well-characterized synthetic vaccines, which are created via the bioconjugation of the antigen to a carrier protein or virus-like particle (VLP), elicit more reliable and robust responses, and process chemistry enhancements address batch variability issues (158). Antigens targeted by vaccines include peptides, carbohydrates, and small molecules. Immunopharmacotherapy is a vaccination strategy that aims to remove drugs of abuse from the bloodstream before they enter the CNS. Nicotine (159, 160), cocaine (161), and heroin (162) are just some of the drugs that have been targeted using rationally designed bioconjugate vaccines. Immunopharmacotherapy provides an additional therapeutic modality in the fight against the opioid crisis (163). Fully synthetic vaccines are also under investigation as these would provide the most homogeneous constructs and thus predictable responses in patients. For example, a tumor-associated carbohydrate antigen, T-helper epitope, and TLR2 agonist adjuvant were combined recently to create an anticancer vaccine candidate (164).
Another method of introducing the antigen is via the injection of RNA or DNA that encodes the protein target. Oligonucleotide-based vaccines, therefore, avoid some of the complexities of carrier protein bioconjugation chemistry and can be developed very rapidly at the point of care, a feature that is becoming increasingly important to stay one step ahead of evolving pathogens (165). However, RNA/DNA-based vaccines often suffer from poor delivery and in vivo stability issues. Medicinal chemistry has been used to successfully enhance the performance of oligonucleotide vaccines by improving metabolic stability and packaging of the nucleic acid polymer into nanoparticles to augment cellular delivery (Section 3.7) (166).
Monoclonal antibodies have also benefited from advances in the rational design and synthesis of antigens, particularly for difficult-to-target epitopes. Low-expressing membrane proteins, which also possess limited surface-exposed extracellular domains, often thwart the elicitation of a strong and functionally relevant immune response from cell-based immunization protocols. Synthetic constrained peptides have been used to mimic the conformations of extracellular domains, particularly for discontinuous epitopes, thus focusing the immune response to the generation of functional antibodies and avoiding immunogenic decoy epitopes that may be present in membrane protein targets (167, 168).
Antibodies also enable tissue/cell targeting when conjugated to small molecule drugs. Antibody–drug conjugates (ADCs) selectively deliver a “payload,” often a highly cytotoxic compound, to cancer cells that express a specific epitope (169). The drug is released either via catabolism of the mAb or through a mechanism that relies on a cleavable linker. For instance, upon antigen-mediated internalization, the lower pH of the resulting lysosome causes site-specific hydrolysis of hydrazone-linked payloads (170).
3.6 Peptide and Protein Medicinal Chemistry
Amino acid chemical diversity delivers the highly specific functionality and molecular interactions of peptides and proteins. However, peptides and proteins often possess poor stability in vivo due to the action of peptidases, and their limited cellular permeability (unless enhanced through the incorporation of positively charged cell-penetrating motifs (171)) hinders their development as drugs. Bioconjugation of polyethylene glycol (PEG) enhances peptide and protein stability by masking the drug from proteolytic degradation and reduces potential immunogenicity that could lead to the development of antidrug antibodies (172). Medicinal chemistry optimization is often required to ensure that the PEG group does not negatively impact target engagement or therapeutic protein functionality. Alternatively, a cleavable linker may be incorporated, such as that used for ADCs described above (170). Unnatural amino acids may be encoded into the protein, or synthesized chemically in the case of a smaller peptide, which enables site-specific bioconjugation. For instance, an alkyne-tethered amino acid allowed for the site-specific attachment of azido-PEG using CuAAC click chemistry (173).
Cystine-knot peptides (CKPs, or knottins) have emerged as interesting pharmacological modulators with considerable therapeutic utility. CKPs are approximately 30 amino acids in length containing internal disulfide cystine bridges arranged in a knot conformation. The cystine knot confers impressive metabolic and chemical stability to the peptide, and their oral bioavailability is improved over linear congeners (174, 175). Therapeutically relevant targets of CKPs reported in the literature include ion channels, proteases, integrins, and GPCRs (176).
Although little work has elucidated the mechanisms by which CKPs enter cells, a recent study showed that a fluorescently tagged derivative of the Ecballium elaterium trypsin inhibitor II (EETI-II, a model CKP) is taken up by active endocytosis, resulting in lysosomal accumulation (177).
Many other medicinal chemical biology technologies have been developed to enhance the cellular delivery, bioavailability, and metabolic stability of peptide therapeutic candidates, and these have been reviewed elsewhere. I point the reader to other constraining strategies employing stapling (178) and bicyclization (179, 180) as representative approaches that illustrate the value of rational molecular design and synthesis that have been shown to enhance the peptide modality (181). More studies are required to further elucidate the mechanisms of cellular distribution and the relationships between peptide/protein structure and internalization that will significantly advance the field.
3.7 Oligonucleotide Therapeutics
Several oligonucleotide antisense drugs have been approved in recent years (182). Synthetic antisense nucleic acids bind to, and inactivate, target mRNA with high specificity (183). Oligonucleotide generally possesses poor bioavailability with no cellular permeability due to the highly charged nature, and size, of the oligomer. They also have high metabolic instability resulting from facile cleavage of the phosphodiester backbone by nucleases. Chemical modifications of the nucleobases, phosphodiester linkages, or sugar moieties have addressed the delivery and stability issues of oligonucleotide therapeutics (Figure 17). For instance, nucleic acid analogs replace the charged phosphate linker with phosphorodiamidate morpholine-containing linkages, and the resulting oligomer possesses improved nuclease stability (184). Locked nucleic acids (LNAs) are modified RNAs where a methylene bridge between the 2′ oxygen atom and 4′ carbon enhances antisense affinity for mRNA through improved backbone preorganization, and nuclease stability is also significantly augmented (185). Other oligonucleotide therapeutic modalities such as double-stranded small interfering ribonucleic acid (siRNA) that catalytically degrades target mRNA have also been enhanced through medicinal chemical biology. Conjugation with trivalent N-acetylgalactosamine (GalNAc) enables siRNA delivery to the liver via binding of the sugar motif to the asialoglycoprotein receptor on hepatocytes, triggering endocytosis of the therapeutic (186, 187). 5′-exonuclease was found to be the prevalent siRNA degrading enzyme in endo-lysosomes and stabilization of the 5′ regions of the siRNA strands significantly increased metabolic stability of the oligonucleotide in vivo (188).

Aptamers are oligonucleotide species selected and enriched from combinatorial libraries to bind various target molecules such as drugs and proteins (189, 190). Pegaptinib targets VEGF for macular degeneration and was the first clinically approved aptamer. Pegaptinib is PEGylated to enhance its pharmacokinetics, as for peptides described above. A recent advance in the area was the creation of PEGylated aptamer libraries to expedite the discovery of oligonucleotides with improved bioavailability and the development of direct in vivo selection methods (191, 192).
The literature concerning the medicinal chemistry optimization of oligonucleotides is vast. I have highlighted some important advances in the field, but recent reviews of the area provide considerably greater detail for the reader (193-195).
3.8 Small and Large Molecule Combinations
Often, small and large molecule therapeutics are compared in debates regarding which may be the better approach for a particular indication (196, 197). However, the ability to combine small and large molecule modalities provides exciting opportunities to enhance therapeutic efficacy. Factor Xa is a serine endopeptidase that plays a key role in the coagulation cascade. Small molecule inhibitors of FXa enzymatic activity such as rivaroxaban have proven therapeutic utility as anticoagulation medicines (198), although modalities with enhanced efficacy are required. An RNA aptamer inhibitor of factor Xa (FXa) was developed that was found to bind outside the catalytic site of the enzyme (Figure 18), thus preventing interaction with a protein cofactor (199, 200). As a result, combination of the aptamer (11F7t) with rivaroxaban prevented clotting in human blood more effectively than either agent alone (199).

Such factor Xa small molecule blood thinners also possess risks from overdosing. A modified FXa decoy protein was developed as an antidote to treat major bleeding episodes (201). This work is an interesting example where small and large molecule drug discovery meets to address patient risks.
The potential for enhancing checkpoint inhibitor mAb therapy with small molecule combinations has been highlighted, and many clinical trials are currently exploring these opportunities (202). Vaccines are increasingly being augmented through the addition of small molecule adjuvants such as toll-like receptor agonists with improved side-effect profiles (203).
Other examples include the use of small molecules that potentiate the immunogenicity of dendritic cell immunotherapies (204, 205) and rapalogue mTOR inhibitors that improve influenza vaccine efficacy in the elderly via immunosenescence amelioration (206). Small molecules are even being used to improve CRISPR-Cas9 gene-editing efficiency (207). A deeper appreciation of pathomechanisms and potential therapeutic modalities will enable the medicinal chemical biologist to rationally select combinations with the best efficacy and safety in the future.
4 Conclusions
This article has highlighted some of the emerging opportunities in medicine research that lie at the interface of chemistry and biology. Medicinal chemical biology should improve our odds of identifying the best therapeutic target and progressing the best therapeutic modality. I have described how chemical biology technologies can be used to identify new therapeutic targets and how the availability of a broad palette of both small and large molecule modalities enables agnostic drug discovery strategies. Therapeutic development will be informed by advances in “omics,” next-generation sequencing, and bioinformatics, particularly where extensive profiling of existing drugs will enable broader repurposing efforts and combinatorial regimens. Synthetic biology will continue to drive many of the advances being made in the area (e.g. codon reassignment, click chemistry, DNA-encoded libraries, and genomic manipulation). Additional future developments include the creation of point-of-care precision vaccines (208) and even synthetic nanomachines that can detect and kill cancer cells (209).
As discussed, medicinal chemical biology will continue to advance our understanding of disease and play an increasingly important role in the selection of therapeutically relevant targets. The four pillars of successful target validation (proof of exposure at the site of action, measurement of target engagement, proof of functional pharmacology, and perturbation of a relevant phenotype – see Figure 2 (5)) are relevant for all therapeutic modalities and indeed essential where concerns exist regarding adequate delivery to the site of action (such as intracellular targets of a peptide or high molecular weight PROTACs).
The positive cultural influences that are intrinsic to chemical biology research are worthy of mention; the traditional boundaries between separate disciplines are blurred, as are the boundaries between academic and industrial research. In this environment, the focus becomes defining the right question and selecting, or developing, the right approach to answer that question in an unbiased manner. Medicinal chemical biologists are ideally placed to facilitate these aims, and they should be provided with the autonomy to deliver on the promise of chemical biology in the drug discovery setting. A lack of research at the interface of physical and life sciences and a paucity of truly collaborative endeavors has hindered therapeutic innovation in pharmaceutical R&D, and medicinal chemical biology should be fostered to address these issues.
That said, over the past 15 years or so, I have seen a growing appreciation within the industry of the value of chemical biology. Some large companies have embraced the discipline, and a considerable percentage of their research organizations now effectively work at the chemistry–biology interface. Many new small companies have been seeded by innovations in chemical biology, which has generated great excitement and investment in the biotech sector. The future for medicinal chemical biologists would appear to be very bright.
Disclaimer
LHJ receives funding from Deerfield.

