

Canavan disease: Clinical features and recent advances in research
Abstract
Canavan disease (CD) is a genetic neurodegenerative leukodystrophy that results in the spongy degeneration of white matter in the brain. CD is characterized by mutations in the gene encoding aspartoacylase (ASPA), the substrate enzyme that hydrolyzes N-acetylaspartic acid (NAA) to acetate and aspartate. Elevated NAA and subsequent deficiency in acetate associated with this disease cause progressive neurological symptoms, such as macrocephaly, visuocognitive dysfunction, and psychomotor delay. The prevalence of CD is higher among Ashkenazi Jewish people, and several types of mutations have been reported in the gene coding ASPA. Highly elevated NAA is more specific to CD than other leukodystrophies, and an examination of urinary NAA concentration is useful for diagnosing CD. Many researchers are now examining the mechanisms responsible for white matter degeneration or dysmyelination in CD using mouse models, and several persuasive hypotheses have been suggested for the pathophysiology of CD. One is that NAA serves as a water pump; consequently, a disorder in NAA catabolism leads to astrocytic edema. Another hypothesis is that the hydrolyzation of NAA in oligodendrocytes is essential for myelin synthesis through the supply of acetate. Although there is currently no curative therapy for CD, dietary supplements are candidates that may retard the progression of the symptoms associated with CD. Furthermore, gene therapies using viral vectors have been investigated using rat models. These therapies have been found to be tolerable with no severe long-term adverse effects, reduce the elevated NAA in the brain, and may be applied to humans in the future.
Canavan disease (CD) is an autosomal-recessive progressive neurodegenerative disease that belongs to a group of genetic disorders recognized as leukodystrophy. CD is neuropathologically characterized by the swelling and spongy degeneration of white matter in the brain. CD was first reported by Canavan in 19311 and was identified as a distinct disease by Bertrand and Van Bogaert in 1949.2 Although CD has been reported in communities throughout the world, it was shown to be more prevalent in Ashkenazi (Eastern European) Jewish people.3 The disease has been attributed to a deficiency in aspartoacylase (ASPA) activity. ASPA is a zinc carboxypeptidase enzyme that is responsible for the breakdown of aspartic acid or N-acetylaspartic acid (NAA), the absence of which results in the accumulation of NAA in the brain. ASPA is normally found in oligodendrocyte progenitor cells and oligodendrocytes in the brain, with smaller amounts being reported in microglia and brainstem neurons. NAA, one of the most prevalent small molecules in the brain, is hydrolyzed to acetate and aspartate in oligodendrocytes.4
Many researchers initially believed that the high NAA associated with CD led to the impeded production of myelin and subsequent spongy degeneration in the brain,5, 6 but this is now being disputed. Cloning of the human ASPA gene has enabled molecular genetic studies of CD.3
Clinical course of CD
Three clinically distinct groups of CD have been identified: (i) the congenital form with severe symptoms in the first few weeks of life; (ii) the infantile form, the most common form in which the disease is apparent by 6 months of age; and (iii) the juvenile form, in which the disease is apparent by the age of 4 or 5.5 Infants with CD typically appear to be normal in the first few months of life. Early signs of CD include irritability and hypotonia with poor head control. The common symptoms of CD include head lag, macrocephaly, hypotonia, ataxia, inadequate visual tracking, poor sucking ability, and intellectual disabilities.7, 8 In many cases, developmental delays and macrocephaly become noticeable after 6 months of age. In spite of profound delays, CD patients can sometimes interact with others, smile, and reach for objects. CD patients later develop optic atrophy, and hypotonia of the arms and legs converts to limb stiffness and spasticity, and axonal hypotonia persists. These patients become increasingly debilitated with age, often having seizures and being unable to move or swallow voluntarily. The long-term prognosis of a typical CD case is still poor; death typically occurs before adolescence, while some patients with milder forms survive beyond their second decade of life.
Clinical examination for diagnosis
The specific method currently used to diagnose CD is urine testing.9 CD is caused by a deficiency in ASPA, which hydrolyzes NAA to aspartate and acetate in the brain. Therefore, urinary NAA is markedly higher in CD patients, often more than 100-fold, than in normal individuals. Slightly elevated NAA (approximately 4–6-fold) have been reported in other cases of leukodystrophy.10, 11 Therefore, this diagnostic procedure is accurate for the screening and chemical diagnosis of CD, with a good cost–benefit ratio. Genetic testing for the ASPA gene mutation can also lead to a definite diagnosis of CD. Cultured skin fibroblasts were previously shown to manifest this enzyme deficiency, even in the absence of an ASPA gene mutation.3, 12 Microscopy shows spongy degeneration throughout the white matter, demonstrating vacuole formation in the myelin sheaths, astrocyte swelling, and deformed mitochondria.
In neurophysiological examinations, electroencephalograms can be diffusely slow with paroxysmal features. Evoked potentials are often delayed or absent, whereas the nerve conduction velocity is typically normal.
Neuroimaging of CD
Computed tomography of the head has shown diffuse hypodensity in the white matter of the brain, while magnetic resonance imaging (MRI) showed diffuse cerebral white matter degeneration. The most severe abnormalities are present in the subcortical white matter with a mildly swollen aspect, and central white matter structures, such as the periventricular rim of the white matter and the internal capsule, are generally preserved. The central white matter also becomes involved as the disease progresses, and white matter atrophy has been reported. The globus pallidus and thalamus are often involved, whereas the putamen and caudate nucleus are spared, which is characteristic of CD (Fig. 1).13 Nuclear magnetic resonance spectroscopy (MRS) has shown that NAA is higher in the brains of CD patients than in those of normal individuals (Fig. 2).14

Chronological features of magnetic resonance imaging (MRI) in a female Japanese Canavan disease patient at (a–d) 15 years and (e,f) 25 years of age. (b,e) T1 and (a,c,d,f) T2-weighted MRI show the involvement of the diffuse white matter, including the corpus callosum and internal capsule, as well as the ( ) globus pallidus. The (
) globus pallidus. The ( ) putamen and (
) putamen and (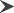 ) caudate nucleus were spared. There are signs of diffuse cerebral and cerebellum atrophy.
) caudate nucleus were spared. There are signs of diffuse cerebral and cerebellum atrophy.

Features of 1H magnetic resonance spectroscopy in a female Japanese Canavan disease patient (same as in Fig. 1). The highly elevated N-acetylaspartic acid (NAA)/choline ratio (6.1; normal range, 1.0–2.4) was characteristic.
Differential diagnosis
Macrocephaly has been reported in Alexander disease, Tay–Sachs disease, and other neurodegenerative diseases. Hydroxymethylglutaric aciduria also leads to macrocephaly and involvement of the white matter. Spongy degeneration of the brain can occur with viral encephalitis, mitochondrial disease, and other metabolic diseases.
Molecular basis of CD
The human ASPA gene, which is localized on the short arm of chromosome 17 (17p13-ter), was cloned by Kaul et al. in 1991.15 The human ASPA gene spans 30 kb and contains five introns and six exons. Over 96% of CD patients among Ashkenazi Jewish populations have either of two mutations. One is a missense mutation in codon 285, which causes glutamic acid to be substituted with alanine (Glu285Ala). The other is a nonsense mutation on codon 231, which converts tyrosine to a stop codon (Tyr231X). The carrier frequency of these two mutations among Ashkenazi Jewish populations has ranged from 1:37 to 1:40, with a prevalence rate of 1 per 6400–13500 live births.4, 16
Mutations are different and more diverse in non-Jewish patients. The most common mutation in non-Jewish patients, a missense mutation that substitutes alanine for glutamic acid (Ala305Glu), has been detected in codon 305.17
More than 50 mutations have been identified in the human ASPA gene, most of which are single base pair changes in the coding region that typically result in the loss of ASPA enzymatic activity. While all patients in whom mutations have been detected have exhibited psychomotor limitations, their onset and severity varied depending on the specific mutation.18, 19
Japanese case of CD
Our questionnaire survey identified only one CD patient in Japan, and this is also the only case that has been reported with the missense mutation I143T.20-22 The patient, a woman, is now 26 years old. She had macrocephaly, gross motor development retardation, and hypotonia since early infancy. She was diagnosed with CD at the age of 4. She could not sit alone due to slowly progressive spastic tetraplegia, but was relatively frequently able to attend school. She had difficulty swallowing at the age of 17 because of progressive bulbar paralysis, and was subsequently fed via a nasogastric tube. She underwent laparoscopic anti-reflux surgery for gastroesophageal reflux disease at the age of 20. Given that the bulbar paralysis is not currently considered to be severe, she does not require any respiratory devices. Although the frequency of tonic seizures increased after the age of 25, levetiracetam has effectively reduced the number of seizures experienced. Figure 1 shows MRI done at the ages of 15 and 25. Subcortical-dominant diffuse white matter was found to be involved. The anterior part of the corpus callosum was highly degenerated. The bilateral globus pallidus and thalamus were also involved, whereas the putamen and caudate nucleus appeared to be spared. Although there are signs of diffuse brain atrophy including the cerebellum, the size of the brainstem has been preserved.
Figure 2 shows MRS of a white matter region at the age of 15. Consistent with previous studies, high NAA concentration and elevated NAA/choline ratio were found to be characteristic in the Japanese patient. Figure 3 shows the 99mTc-ethyl cysteinate dimer single-photon emission computed tomography done at the age of 15. A frontal predominant decrease in cerebral blood flow was observed.

Possible pathological mechanism responsible for CD
N-Acetylaspartic acid appears to be synthesized exclusively in neurons and has been isolated from mitochondrial and microsomal fractions.23 NAA and its related dipeptide N-acetyl-aspartyl-glutamate (NAAG) are transported from cytoplasm to the extracellular space by transporters, and NAA is taken up by oligodendrocytes through a dicarboxylic acid transporter prior to being hydrolyzed by ASPA (Fig. 4).

Schematic representation of N-acetylaspartic acid (NAA) synthesis in neurons and degradation in oligodendrocytes. ACS, acetyl CoA synthetase; ASPA, aspartoacylase; AspNAT, aspartate N-acetyltransferase; NAAG, N-acetyl-aspartyl-glutamate; PDH, pyruvate dehydrogenase; TCA, tricarboxylic acid.
N-Acetylaspartic acid serves as a clinical marker of the neuronal metabolic integrity of the brain. In contrast with the decrease reported in NAA in many other neurodegenerative diseases, CD is unique because it is associated with elevated NAA in the brain. A marked rise of NAAG concentration has also been reported in patients with a Pelizaeus–Merzbacher-like syndrome, in which there is an absence of myelin.24 Establishing why increases in NAA or NAAG lead to white matter degeneration or dysmyelination is very important, but the precise function of NAA in the development of the central nervous system (CNS) remains unknown.
Several hypotheses have been proposed to explain the pathophysiology of CD in the CNS.
First, demyelination may reflect the direct action of NAA on oligodendrocyte NMDA receptors. No current, however, was evoked by NAA in oligodendrocytes in a rat study. Therefore, the action of NAA or NAAG on oligodendrocyte NMDA receptors is unlikely to be a major contributor to white matter damage.25
Second, NAA may serve as a molecular water pump to remove metabolic water from mitochondria and neurons through its hydrolysis into acetate and aspartate by ASPA. This hypothesis corresponds to astrocytic edema and the formation of vacuoles in CD as a result of the accumulation of NAA.26 A previous study, however, showed that NAA was non-toxic even at high concentration,27 and no functional improvement was reported in CD mice even after NAA decreased due to the expression of an introduced normal ASPA gene.28 An immunohistochemical study of the Nur7 mouse model of CD showed that aquaporin 4 (AQP4) was located throughout the cytoplasm in CD mice, but it was located exclusively in the astrocytic end-feet in control mice. This indicates that the astroglial regulation of water homeostasis may be involved in the partial prevention of spongy degeneration, and AQP4 may be a potential therapeutic target for CD.29
Third, NAA may be essential for lipid synthesis and myelination in the CNS during the period of postnatal myelination.12 CD is characterized not only by an increase in NAA but also by a decrease in acetate and aspartate,12, 30, 31 and a reduction in free acetate for lipid synthesis subsequent to the loss of ASPA function may contribute to the disease etiology. Spongiform degeneration in CD brains has been attributed to the failure of NAA to act as an acetate carrier from mitochondria to the cytosol, leading to impaired lipogenesis.32, 33 Therefore, one of the main causes of CD may be a decline in acetyl groups in the absence of ASPA activity. Non-polar and polar lipid levels, critical for myelin synthesis, were found to 21–38% lower in ASPA knockout mice than in wild type, whereas other lipids were not altered significantly.32 Moreover, a reduction of cerebroside and sulfatide, component glycolipids of myelin in the white matter, was also reported both in the rat CD model and in human CD patients. The reduction observed in lipid level, however, may not have directly correlated with the clinical severity of the disease. These results suggest that the pathogenesis of CD is not restricted to a deficiency of acetate.
Fourth, NAA may play an important role in maintaining the metabolic integrity of oligodendrocytes. Elevation in oxidative stress markers was shown to precede the loss of oligodendrocytes and demyelination in the early days following birth.34
Last, besides the role of acetate in myelin formation and maintenance, acetylation also modulates the function of nucleosomal histones, which are components of chromatin. Therefore, a decrease in acetate may alter the expression of genes considered to be important for the maturation of oligodendrocytes.35 Although NAA is produced and localized primarily in neurons,36 high NAA concentration has also been reported also in immature oligodendrocytes. NAA, however, was not detected in mature oligodendrocytes or astrocytes, which suggested the important function of ASPA in immature oligodendrocytes.37 In rat cortical cultures, the presence of ASPA activity as well as the expression of ASPA mRNA and protein have been reported in non-myelinated oligodendrocytes.31, 38, 39 Although the direct uptake of NAA by oligodendrocytes has not yet been reported, axonal transport of NAA has been demonstrated.40 These findings suggest that the maintenance of myelin is impaired in the absence of NAA-derived acetate. ASPA plays a critical role in the maturation of oligodendrocytes and has also been shown to contribute to the pathophysiology of CD.41 Furthermore, in an adult ASPA knockout mouse study, disruptions have been observed in cell cycle regulation, the acetylated state of nuclear histones, and continuous neurogenesis in neural cell progenitors, as well as severe reduction in certain myelin proteins.42 ASPA may be involved in the epigenetic regulation of myelin maturation and maintenance through the supply of acetate.
Therapeutic approaches to CD
A therapy that affects the progression of CD has not yet been established. Seizures need to be controlled by anticonvulsants. Patients with CD may need nasogastric feeding or feeding by gastronomy. Acetazolamide was found to be beneficial in reducing intracranial pressure, but did not reduce white matter swelling or NAA level.
Dietary supplementation is one of the non-invasive therapies that have been positively correlated with improvement in NAA level in CD patients. Lithium citrate decreased whole-brain NAA in both rat models and human subjects.43 More recent studies noted improved scores in gross motor functioning and visual tracking in CD patients treated with lithium citrate from an early age compared to untreated control groups of CD patients.44 Acetate supplementation represents another potential therapy for CD that is easy and inexpensive.45 Oral treatment with glyceryl triacetate (GTA) in CD mice led to a 17-fold increase in acetate in the brain and improved motor function, while NAA level in the brain was not significantly increased.46 This therapy is now being used in CD patients. Although high-dose GTA (up to 4.5g/kg per dose) treatment in CD infants resulted in no improvement in their clinical status, no significant side-effects or toxicity were observed. The importance of earlier intervention has been suggested.47 Another possible supplement is triheptanoin, an odd-carbon triglyceride, which is a dietary anaplerotic substrate that provides ketone bodies capable of traversing the blood–brain barrier and increasing the mass of tricarboxylic acid (TCA) cycle intermediates. Triheptanoin was shown to be effective in the treatment of mitochondrial oxidation and pyruvate metabolism.48, 49 A previous study reported that interventions with triheptanoin therapy reduced oxidative stress, promoted long-term survival of oligodendrocytes, and increased myelin in the brain in the nur7 mouse model.50 The novelty of that study lies in the potentially anaplerotic substrate, with the aim of supporting TCA cycle oxidative integrity in addition to fatty acid synthesis during developmental myelination. The early provision of triheptanoin as an alternative energetic substrate to the nur7 mouse model promoted myelination by reducing the metabolic demands placed on the oxidation of glucose by fatty acid synthesis.50 NAA was found to be higher in the brains of neonatal triheptanoin mice than in control mice, and triheptanoin had no effect on NAA. NAA is known to have a negative impact on anti-oxidant defenses; therefore, antioxidants may have therapeutic application in CD patients.51
Several studies recently attempted to use gene therapies for the treatment of CD. A therapy using adeno-associated virus (AAV) was used in the tremor rat, a genetic model of CD, and ASPA activity was subsequently detected in the CNS neurons of this rat. Although NAA was also reduced in the brain, motor functions remained unimproved.28
The application of gene therapy for CD currently faces several challenges. The varied rates of disease progression and small number of patients have confounded any interpretation of the effects of this therapy. Moreover, it is difficult to attempt a cohort study of age-matched or similar-phenotype patients due to variations in the mutations that cause CD.
Leone et al. reported the findings of a long-term follow up of gene therapy with an AAV vector carrying the ASPA gene (AAV2-ASPA) in 13 CD patients.52 Each patient received 9 × 1011 vector genomes via intraparenchymal delivery at six brain infusion sites. The gene therapy was tolerable, no severe long-term adverse effects were noted, elevated NAA in the brain was reduced, the progression of brain atrophy was slowed, and improvements were observed in the frequency of seizures.52 Moreover, neurological examination showed significant improvement in motor functions in younger cohorts of treated CD patients, indicating the possible advantage of early therapeutic intervention.53
Conclusion
In addition to the accumulated medical research on CD, parent and family community support has been increasing. Internet forum or family websites are also meaningful for CD patients.19 The prognosis of childhood CD has gradually improved due to advances in comprehensive care. Future research will hopefully facilitate development of a safe and therapeutic approach for CD patients and improve their quality of life.
Acknowledgments
This study was supported by the Health and Labor Sciences Research Grants for Research on Intractable Diseases, the Ministry of Health, Labor and Welfare.

