

Lipids and adipocytes involvement in tumor progression with a focus on obesity and diet
Funding information: This work was supported by an Italian Ministry of Health Grant for the project GR-2019-12370014.
Summary
The adipose tissue is a complex organ that can play endocrine, metabolic, and immune regulatory roles in cancer. In particular, adipocytes provide metabolic substrates for cancer cell proliferation and produce signaling molecules that can stimulate cell adhesion, migration, invasion, angiogenesis, and inflammation. Cancer cells, in turn, can reprogram adipocytes towards a more inflammatory state, resulting in a vicious cycle that fuels tumor growth and evolution. These mechanisms are enhanced in obesity, which is associated with the risk of developing certain tumors. Diet, an exogenous source of lipids with pro- or anti-inflammatory functions, has also been connected to cancer risk. This review analyzes how adipocytes and lipids are involved in tumor development and progression, focusing on the relationship between obesity and cancer. In addition, we discuss how diets with varying lipid intakes can affect the disease outcomes. Finally, we introduce novel metabolism-targeted treatments and adipocyte-based therapies in oncology.
Abbreviations
-
- AA
-
- arachidonic acid
-
- ASC
-
- adipose stromal cell
-
- AT
-
- adipose tissue
-
- ATP
-
- adenosine triphosphate
-
- BMI
-
- body mass index
-
- CAA
-
- cancer-associated adipocyte
-
- CAD
-
- coronary artery disease
-
- CAF
-
- cancer-associated fibroblast
-
- CPT1
-
- carnitine palmitoyltransferase 1
-
- CPT-1
-
- carnitine palmitoyltransferase-1
-
- CR
-
- caloric restriction
-
- CTC
-
- circulating tumor cell
-
- DDS
-
- drug delivery system
-
- DHA
-
- docosahexaenoic acid
-
- DHA
-
- docosahexaenoic
-
- ECM
-
- extracellular matrix
-
- EMT
-
- epithelial-mesenchymal transition
-
- EPA
-
- eicosapentaenoic
-
- EV
-
- extracellular vesicle
-
- FA
-
- fatty acid
-
- FAO
-
- fatty acids oxidation
-
- FASN
-
- fatty acid synthase
-
- FFA
-
- free fatty acid
-
- FMD
-
- fasting-mimicking diet
-
- HCC
-
- hepatocellular carcinoma
-
- HFD
-
- high-fat diet
-
- LBCL
-
- large B-cell lymphoma
-
- LCFA
-
- long-chain fatty acid
-
- LD
-
- lipid droplet
-
- LDLR
-
- low-density lipoprotein receptor
-
- MSC
-
- mesenchymal stem cell
-
- mTOR
-
- mammalian target of rapamycin
-
- MUFA
-
- monounsaturated fatty acid
-
- NK
-
- natural killer
-
- NP
-
- nanoparticle
-
- NSCLC
-
- non-small cell lung cancer
-
- OXPHOS
-
- oxidative phosphorylation system
-
- PDX
-
- orthotopic-patient-derived xenograft
-
- PUFA
-
- polyunsaturated fatty acid
-
- ROS
-
- reactive oxygen species
-
- SAT
-
- subcutaneous adipose tissue
-
- SCFA
-
- short-chain fatty acid
-
- SFA
-
- saturated fatty acid
-
- TAG
-
- triacylglycerol
-
- TCA
-
- tricarboxylic acid cycle
-
- TG
-
- triglyceride
-
- TGF
-
- transforming growth factor
-
- TME
-
- tumor microenvironment
-
- TNBC
-
- triple-negative breast cancer
-
- VAT
-
- visceral adipose tissue
-
- WD
-
- Western diet
1 INTRODUCTION
The prevalence of obesity has increased worldwide over the last decades.1 The obesity state refers to an accumulation of excess fat deposits with a body mass index (BMI) over 30 kg/m2.2 In individuals with obesity, genetic and environmental factors, such as food contaminants, as well as eating habits, play an important role in determining body composition.2 Additionally, an imbalance between calorie intake and physical activity results in a chronic positive energy balance, leading to weight gain.3
According to numerous epidemiological studies, obesity is related to an increased risk of developing certain cancers, such as pancreatic, colorectal, postmenopausal breast, ovarian, endometrial, liver, kidney (renal cell), esophagus (adenocarcinoma), gastric, and gallbladder.4-6 A high BMI is also linked to a more aggressive disease and adverse outcomes in prostate cancer. This might be linked to the migration of prostate cancer cells towards periprostatic adipose tissue (AT), which provides lipid mediators and inflammatory cytokines in support of cancer cell growth and motility.7, 8
However, regardless of global body composition, fat depots can be divided into visceral and subcutaneous, which have different structures and functions in cancer development and progression. Visceral adipose tissue (VAT) has been linked to increased amounts of tumor-promoting metabolites, including free arachidonic acid, phospholipases, prostaglandin synthesis-related enzymes, and lipid metabolites related to inflammation compared to subcutaneous adipose tissue (SAT).9 VAT adipocytes are also lipolitically more active than SAT adipocytes and contribute more to plasma-free fatty acid levels, particularly in obese individuals.10 In patients with colon, esophageal, and renal cancers, VAT has been linked to worse clinical outcomes, which include short- and long-term survival, recurrence, and postoperative.9, 10 Instead, research exploring the prognostic significance of subcutaneous obesity in cancer patients has produced inconsistent findings. For instance, a high SAT was linked to a higher survival rate in patients with bone metastases and hepatocellular cancer.9 Hence, there is growing interest in understanding how adipocytes and AT contribute to tumor development and progression via direct and indirect crosstalk with cancer cells, considering that adipocytes are not always in immediate contact with cancer cells.
In addition, an increasing body of evidence highlighted the role of metabolism in tumors. In particular, dysregulated lipid metabolism is a feature of malignant cells that can exploit cells in the tumor microenvironment (TME) to enhance their use of lipids and boost their proliferation.11 Indeed, cancer cells can increase de novo lipogenesis and lipid accumulation, fatty acid (FA) uptake, and FA oxidation (FAO) for energy production.12-14 In particular, the presence of cancer-associated adipocytes (CAAs) has been found in the TME of breast and abdominally metastasizing cancers (e.g., gastric, colon, and ovarian).15
Other distinctive hallmarks of obesity are low-grade inflammation,16 hormonal alterations,17 insulin resistance, hypercholesterolemia,18 and an intensified adipokines secretion.19 These events can trigger tumorigenesis, cancer progression, and a modified response to therapies18, 20 by reinforcing tumor-promoting inflammation21 and increasing angiogenesis.22
Finally, diet is the source of exogenous lipids and is linked to several human cancers.23 Epidemiological, clinical, and laboratory evidence have linked excess calorie consumption with increased cancer risk, while a low-carbohydrate and low-fat diet is associated with reduced cancer incidence.24, 25 An explanation for this phenomenon could be that dietary alterations may modify circulating concentrations of metabolites, which then influence the composition of the TME, thus driving tumor progression and altering therapeutic responsiveness.26
Our review analyzes how adipocytes contribute to the tumor progression and how lipid metabolism is altered in cancer, with a particular focus on the role of TME. The importance of lipids in cancer will also be analyzed for their function in tumor suppression through ferroptosis, a recently discovered programmed cell death event. We also discuss scientific studies examining the influence of diets with different lipid intakes on disease outcomes. Finally, we analyze the most recent and relevant treatments, which have been developed to target lipid metabolism and adipocyte-based cell therapies in oncology (Figure 1).

2 TUMOR METABOLISM
2.1 Overview
Cancer cells have a prevalent mechanism of producing energy, which differs from normal cells. They use aerobic glycolysis to produce adenosine triphosphate (ATP), and most of the generated pyruvate is converted into lactate. This is unlike normal cells, which use the mitochondrial oxidative phosphorylation system (OXPHOS) to produce energy. This change in metabolism is known as the “Warburg effect,” and it occurs even when oxygen is abundant, despite OXPHOS producing a greater number of ATP molecules.27 By providing precursors for numerous metabolic pathways, the Warburg effect could be advantageous to cancer cells.28 Indeed, substrates for the bioenergetic pathways that support tumor growth, such as nucleotide, lipid, and protein synthesis, including glucose, glutamine, lactate, pyruvate, hydroxybutyrate, acetoacetate, acetate, and free fatty acids (FFAs), are increased in production during cancer.29, 30
2.2 Metabolic coupling between cancer and stromal cells in the TME
Recent findings have uncovered an additional intriguing function of the TME in determining the metabolic profile of tumor cells.31 The TME consists of extracellular matrix (ECM) proteins and several stromal cell types such as endothelial cells, fibroblasts, immune cells, pre-adipocytes, adipocytes, and inflammatory cells. The TME provides nutrients, growth factors, and other extracellular molecules to tumor cells, affecting their metabolism and boosting their growth.32 Recent evidence suggests that tumor cells may exploit stromal cells to get more metabolic substrates such as glucose, lactate, glutamine, and FAs by stimulating their glycolysis and lipolysis pathways to increase energy production through a “reverse Warburg effect.”33 According to the hypothesis provided by Martinez-Outschoorn et al.,29 cancer cells cause oxidative stress in nearby stromal cells, which in turn trigger aerobic glycolysis, thus generating extra lactate and/or pyruvate. These high-energy compounds can then be transported to nearby cancer cells, where they enter the tricarboxylic acid cycle (TCA), causing an increase in oxidative phosphorylation and a boost in ATP generation. The metabolic coupling between stromal and cancer cells was demonstrated in experimental models of breast, ovarian, prostate, and sarcomas.29 This relationship has also been observed in several human tumors, such as prostate,34 lung,35 breast, pancreatic, and gastric cancers.36
The reorganization of metabolic pathways is now recognized as a distinctive feature of malignant cells. It is becoming more and more attractive because of its implication as a potential therapeutic target.37 Currently, several ongoing studies are exploring the development of anticancer treatments that target lactate,38 pyruvate,39 acetyl CoA,40 FAs (discussed in detail in this review),41 and amino acids metabolism.42
2.3 Dysregulated lipid metabolism in support of tumor growth and metastasis
An explanation for the metabolic shift in cancer cells during tumor development is the activation of specific pathways that supply metabolic substrates to support their growth and proliferation. Lipids are among the most important cellular elements whose production increases during cancer progression.39 They are hydrophobic molecules which include sterols, monoglycerides, diacylglycerides, triglycerides (TGs), phospholipids, and glycolipids. The major components of lipids are FAs, a group of molecules consisting of hydrocarbon chains varying in length and saturation.
One of the major functions of lipids is their contribution to composing biological membranes in the form of phospholipids, which are amphipathic molecules.43 Lipids can also act as mediators or signaling molecules.44 The most coherent example consists of polyunsaturated fatty acids (PUFAs), which include arachidonic acid (AA), an omega-6-derived PUFA, and the substrate for the synthesis of eicosanoids, such as prostaglandins, thromboxanes, and leukotrienes. These play a role in tissue inflammation and promote a pro-tumorigenic environment.21, 45 Other PUFAs with signaling functions are the omega-3 FAs (eicosapentaenoic acid [EPA] and docosahexaenoic acid [DHA]), which are believed to reduce inflammatory processes and the risk of breast and other cancers.46 As previously mentioned, tumor cells can promote lipolysis in adjacent adipocytes.11 Accordingly, a metabolic symbiosis takes place between these cells and leads to the storage and mitochondrial use of adipocyte-derived FAs by cancer cells.47 Bochet et al.48 demonstrated that, after co-culture with tumor cells, lipid content is released from tumor-surrounding adipocytes and the FAs are transferred to cancer cells. The movement of lipids has been proven in both prostate49 and ovarian cancers.50 Various studies showed how breast cancer cells exposed to adipocyte-conditioned media, or in co-culture with adipocytes, modified proliferation, migration, and invasion.51, 52 Analogous results have been noticed in 3D cultures53 and xenograft models.54 In particular, Nieman et al.50 proved that primary human omental adipocytes promote gastric, breast, colon, and ovarian cancer cell proliferation and metastasis in vitro and ovarian cancer growth in vivo. They demonstrated that adipocytes facilitate the early homing of ovarian tumor cells to the omentum by producing adipokines. Then, lipid transfer from adipocytes to cancer cells accelerates the growth of metastases. This process may not be specific to ovarian cancer cells and explains the proliferation of other cancerous cell types that migrate to the abdomen or areas with a high concentration of adipocytes.
Lipid metabolism can also be affected by variable oxygen tension and hypoxia in the TME. Tumor cells exposed to hypoxia enhance FA uptake55 and the expression of lipid uptake-associated proteins. In particular, CD36 (a fatty acid transport protein) in the lung, bladder, breast, and ovarian cancers56, 57 and the low-density lipoprotein receptor (LDLR) in pancreatic cancer are associated with unfavorable prognosis in patients.58 Moreover, adverse environmental conditions, such as high acidity59 or glucose deprivation,60 can stimulate the upregulation of FAO, which many tumors rely on for their survival. This has been observed in lung,61 ovarian,50 colorectal,61 and breast62 cancers. Overexpression of FAO enzymes has been observed in these types of cancer,63 and arresting FAO slows down tumor growth in several experimental models. Specifically, inhibiting carnitine palmitoyltransferase 1 (CPT1), the rate-limiting enzyme in FAO, was demonstrated to hinder tumor progression and prolong survival in an orthotopic-patient-derived xenograft (PDX) triple-negative breast cancer (TNBC) model,62 in a PDX model of glioblastoma,64 and in an animal model of liver cancer.65
In an interesting research by Li et al.,66 they examined the role of obesity in metabolic alterations occurring in myeloma patients. By analyzing circulating tumor cells (CTCs) isolated from patients affected by obesity, the authors recognized the involvement of the acetyl coenzyme A (acetyl-CoA) metabolic pathway in myeloma growth. Acetyl-CoA, a key metabolite acting as an essential intermediate for pyruvate to take part in the TCA cycle, is the main precursor of lipid synthesis.67 The limiting enzyme for acetyl-CoA production from acetate is acetyl-CoA synthetase 2 (ACSS2). In metabolic stressful conditions, such as hypoxia and glucose deprivation, ACSS2 can promote cancer progression by acetyl-CoA production.68, 69 Li et al.66 demonstrated that in myeloma patients, the expression of ACSS2 was positively correlated with BMI values. Comparing patients with normal weights (BMI < 25 kg/m2) and patients affected by obesity (BMI > 30 kg/m2), the authors identified many genes involved in pathways related to inflammatory response and metabolic processes, which were differentially expressed in the two groups. Patients with high expression of ACSS2 showed poor overall and relapse-free survival. In general, the patients with BMIs over 30 kg/m2 and high expression of ACSS2 had worse prognosis.66
3 ADIPOCYTES PROMOTE TUMOR PROGRESSION THROUGH LOCAL AND SYSTEMIC MECHANISMS
3.1 Function and endocrine role of adipocytes
Adipocytes play a key role in both growth and metastasis of a number of tumors, including those located in the breast, prostate, ovary, stomach, kidney, and colon.8 These cells are among the constituents of AT, and their hallmark is the ability to store excess calories in the form of triacylglycerols (TAGs) or TGs, which are packaged into large lipid droplets (LDs) and mobilized as needed.70, 71 When energy demand increases, TAGs undergo the process of lipolysis through which they split into their components: FAs and glycerol. Once released, FAs can participate in different pathways, including β-oxidation (or FAO), which generates energy in the form of ATP, re-esterification into TAGs,72 and can act as signaling molecules.73
Adipocytes are also integrated into the systemic metabolism and have a second role as endocrine cells. When the state of positive energy balance is chronic, such as during obesity, the AT changes in adipocyte size, distribution, cellular composition, and function.74 In particular, hypertrophic adipocytes, ectopic fat deposition (such as in the liver or heart), hypoxia, and chronic stress occur, subsequently causing a greater secretion of pro-inflammatory adipokines, which are defined as lipid or protein factors (hormones, cytokines, growth factors, and matrix remodeling components) with endocrine and paracrine functions75 (Figure 2). These include monocyte chemoattractant protein (MCP)-1, tumor necrosis factor (TNF)-α, interleukin (IL)-6, resistin, adiponectin, and leptin.76 Intensified release of these inflammatory mediators induces the chemotaxis of lymphocytes, macrophages, and stromal cells77 towards AT, and this results in low-grade chronic inflammation, a typical feature of obesity, which supports cancer progression.16, 78 For example, TNF-α, IL-6, and MCP-1 are released from adipocytes or from inflammatory cells in AT to stimulate cell proliferation, angiogenesis, and tumor progression.74, 79 Furthermore, these mediators produce reactive oxygen species (ROS) which have mitogenic properties and have been proposed as tumor promoters.80

3.2 Inflammatory adipokines and cytokines in cancer promotion
As reported above, obesity is correlated with a chronic low-grade state of inflammation that is the effect of a heightened concentration of FAs, inflammatory cytokines, immune cells such as macrophages and also adipokines released by adipocytes81 (Table 1).
| Adipokines | Effects on tumor | Affected cancer types | Mechanisms | References |
|---|---|---|---|---|
| Leptin |
(↑) tumor development, angiogenesis and progression (↓) efficacy of anticancer drugs |
Breast |
(↑) cyclin D1, p53, survivin, IL1, E-cadherin, VEGF and its receptor type 2 expression (↑) JAK-STAT3, Akt, JNK, MAPK/ERK1/2, PI3K/AKT-1 Pathways |
82-88 |
| Resistin |
(↑) risk (↑) metastasis |
Gastric, colorectal, liver, pancreatic, ovarian, breast, lung | (↑) EMT via the NF-κB/STAT3 pathway (↑) VEGF production and Akt phosphorylation | 89-98 |
| Adiponectin | (↓) risk | Breast, prostate, gastrointestinal, endometrial, hepatocellular |
(↓) mTOR phosphorylation (↓) production of pro-inflammatory cytokines by inhibiting NF-κB activity (↑) apoptosis |
81, 99-103 |
| IL-6 | (↑) tumor growth, angiogenesis, progression | Esophageal, gastric, colorectal, pancreatic, bladder, breast, ovarian, prostate, hepatocellular, renal cell |
(↑) JAK-STAT3, SH2-SHP-2-Ras-Raf-MEK-ERK and PI3K-Akt pathways |
104-106 |
| TNF-α | (↑) tumor growth, angiogenesis, progression | Lung, pancreatic, breast, prostate | (↑) NF-κB and JNK pathways | 104, 107, 108 |
| MCP-1 | (↑) migration, angiogenesis, tumor development | Colorectal, breast, lung, hepatocellular, pancreatic, renal, nasopharyngeal |
(↑) recruitment of immune cells (↑) PI3K/AKT, MAPK/p38 and JAK/STAT3 pathways |
109-120 |
- Note: The table lists the known adipokines, their effects on different types of tumor and mechanisms of action. (↑): increase; (↓) decrease.
One of the well-known adipokines is leptin, which is produced by local and distant adipocytes and by other cells within the TME and acts in endocrine, paracrine, and autocrine ways.121 Leptin blood levels are directly proportional to body fat mass,122 being overproduced in individuals with obesity. Clinical and experimental studies have proved the role of leptin in supporting tumor development and progression and in decreasing the efficacy of cancer treatments.82 Leptin also has angiogenic properties which may contribute to promoting cancer progression.83 As shown in breast carcinogenesis, leptin signaling regulates a significant number of molecules involved in proliferation, adhesion, migration, invasion, angiogenesis, and inflammation. This occurs through the regulation of the expression of cyclin D1, p53, survivin, IL1, E-cadherin, vascular endothelial growth factor (VEGF), and its receptor type 2, as well as several tissue factors.84, 85 Leptin promotes pro-survival pathways in cancer cells, for example, Janus kinase (JAK)-signal transducer and activator of transcription 3 (STAT3), Akt, and c-Jun N-terminal kinase (JNK) by binding to its receptor ObR.86 Transcription of genes that regulate cellular proliferation and invasiveness, such as telomerase reverse transcriptase, IL-6, transforming growth factor (TGF), matrix metalloproteinases (MMP)9, and MMP13, is induced by the activation of these signaling pathways.87 Moreover, this adipokine cooperates with different oncogenes, cytokines, and growth factors such as JAK/STAT3, mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK)1/2, and phosphoinositide 3-kinase (PI3K)/AKT-1.88 It is interesting to note that leptin signaling also influences metabolic remodeling in cancer cells: in the absence of leptin receptors, mammary cancer cells exhibit increased mitochondrial oxidation and decreased glycolysis.123
Resistin, like leptin, is a type of adipokine that increases the risk of developing a variety of tumors such as gastric,89 colorectal,90 liver,91 pancreatic,92 ovarian,93 breast,94 and lung cancer.95 Resistin mainly promotes epithelial–mesenchymal transition (EMT), a process during which individual cells enhance their motility and acquire an invasive phenotype as a result of loss of epithelial cell junctions, reorganization of their cytoskeleton, and reprogramming of gene expression, events that promote cancer progression.96 Resistin stimulates EMT via the nuclear factor-κB (NF-κB)/STAT3 signaling pathway, VEGF production, and Akt phosphorylation,97 thus stimulating metastasis.98
Adiponectin is an adipokine that is conversely related to body fat mass and it has anticancer effects. Numerous pieces of evidence proved this inverse relationship between adiponectin blood level and cancer risk in breast,99 prostate,100 gastrointestinal,101 endometrial102 cancers, and hepatocellular carcinoma.103 Adiponectin reduces the mammalian target of rapamycin (mTOR) phosphorylation, which in turn limits the production of pro-inflammatory cytokines by inhibiting NF-κB activity and induces apoptosis.81, 103
Similarly, adipocytes produce the cytokines IL-6 and TNF-α that play a key role in inflammation and immunity. Besides, there is increasing evidence that the AT of individuals with obesity has a great infiltration of macrophages that are stimulated by adipocytes to switch from the M2 phenotype to M1. This causes a shift from anti-inflammatory cytokines produced by M2 macrophages, such as IL-10, to pro-inflammatory cytokines such as IL-6 and TNF-α produced by M1 macrophages.124 Therefore, both the production and secretion of these two cytokines rise in individuals with obesity and enhance tumorigenesis via their pro-inflammatory effects. IL-6 and TNF-α intensify tumor proliferation, survival, and angiogenesis driving carcinogenesis and cancer progression.104 TNF-α acts as a tumor promoter also activating the NF-κB and JNK signaling pathways.107 High levels of circulating TNF-α were found in patients with lung, pancreatic, breast, and prostate cancers.108 Likewise, serum IL-6 had been reported to be associated with poor prognosis in oesophageal, gastric, colorectal, pancreatic, bladder, breast, ovarian, prostate, hepatocellular, and renal cell carcinomas.105 This inflammatory factor can activate the JAK-STAT3 pathway, the Src homology 2 (SH2)-containing protein tyrosine phosphatase-2 (SHP-2)-Ras-Raf-mitogen-activated protein kinase (MEK)-ERK pathway and the PI3K-Akt pathway, therefore taking part in differentiation, proliferation, migration, and apoptosis of cancer cells.106
MCP-1, also known as chemokine (C–C motif) ligand 2 (CCL2), is another inflammatory factor secreted by adipocytes. The binding between CCL2 and its receptor CCR2 activates a transduction pathway that leads to the recruitment of immune cells, especially monocytes, memory T lymphocytes, and natural killer (NK) cells towards the inflammatory region, playing critical roles in the immune response.110 Other signaling pathways activated by CCL2 are PI3K/AKT, MAPK/p38, and JAK/STAT3 which play a vital role in anti-apoptosis, and cell migration, leading to oncogenic advancement.111-113 Also, the expression of CCL2 was significantly associated with neo-angiogenesis.114 It has been demonstrated that the CCL2–CCR2 signaling axis participated in the development of different types of cancer, such as colorectal,115 breast,109 lung,116 hepatocellular,117 pancreatic,118 and renal cancers119 and nasopharyngeal carcinoma.120
Even though adipokines may independently affect the development of various obesity-related cancers, they could also act together through the crosstalk of their respective downstream signaling pathways. The link between leptin and some cancers often involves adiponectin as well. In esophageal cancer, for example, leptin-induced proliferation of tumor cells could be arrested by adiponectin through AdipoR1 activation.101 Likewise, leptin-induced proliferation of hepatocellular tumor cells can be inhibited by adiponectin via the STAT3 signaling pathway.74
3.3 Cancer-associated adipocytes
Dirat et al.51 emphasized the crucial role of the peritumoral AT in the progression of breast cancer and were the first to hypothesize that tumor cells may rewire adipocytes in their adjacent microenvironment to assume pro-inflammatory and aggressive phenotypes.125 These peritumoral adipocytes were named cancer-associated adipocytes (CAAs). Notably, they discovered that adipocytes co-cultivated with breast cancer cells exhibit peculiar features.51, 126 CAAs displayed a reduction in size because of the transfer of lipids to cancer cells and an activated phenotype through the downregulation of adipocyte markers like resistin, adiponectin, and fatty acid binding protein (FABP4). Besides, they overproduce adipose-derived factors via endocrine and paracrine signaling pathways, including CCL2, CCL5, IL-6, TNF, VEGF, and leptin.51, 127 CAAs also generate fibroblast-like cells known as adipocyte-derived fibroblasts when there are cancer cells present, especially at the tumor-invasive front. These cells may represent a subpopulation of cancer-associated fibroblasts (CAFs), and, like them, they are engaged in the malignant evolution of tumors.48 Accordingly, Zhu et al.128 found that during tumor growth, adipocytes from tumor-invasive mammary fat de-differentiate into fibroblasts-like progenitor cells and fully integrate into the TME, resulting in an adipocyte mesenchymal transition (AMT). The de-differentiated adipocytes change into a variety of cell types related to ECM remodeling, inflammation, and immune response. Although the de-differentiated cells have different metabolisms from CAFs, they have similar impacts on the proliferation of tumor cells. Moreover, this de-differentiation has also been found in histological studies of abdominally metastasizing cancers (ovarian, renal, pancreatic, gastric, and colon).128
As reported by Attané and Muller,14 CAAs undergo multiple catabolic processes resulting in the release of metabolites, such as lactate, pyruvate, FFAs, and ketone bodies. Additionally, a study by Dirat et al.51 demonstrated that CAAs stimulate the invasive capacities of murine and human breast cancer cell lines. They noticed that when they cultivated tumor cells in a medium conditioned by adipocytes, which have never been co-cultivated with tumor cells, there was no significant increase in the cells' invasive abilities for any of the cell lines. However, when the conditioned medium was derived from adipocytes previously co-cultivated with tumor cells, significant stimulation of tumor invasive capacities was observed in all the tested models compared with cells grown in the control medium. The results highlighted that a cross-talk between the two cell populations is necessary to obtain this effect. The tumor cell-derived signaling molecules induce lipolysis of adipocytes and promote the evolution of adipocytes to CAAs.71 This CAA-activated phenotype has been confirmed in vitro in several tumor histotypes (prostate, lung, ovarian, breast129 and pancreatic cancers,130 melanoma,131 hepatocarcinoma,132 and leukemia129) and in vivo at the invasive front of human breast cancer.51, 133
Besides CAAs, AT provides progenitor cells that may explain the mechanisms involved in the relationship between obesity and cancer progression. These cells, known as adipose stromal cells (ASCs), are a component of the stromal vascular fraction of AT and are found in greater amounts in individuals with obesity.134 They exhibit multipotency and proliferative capacity similar to bone marrow mesenchymal stem cells (MSCs), but they also represent a source of CAFs.135 It has been suggested that the recruitment of ASCs by tumor is enhanced in individuals with obesity and that ASCs promote tumor vascularization, proliferation of malignant cells, and resistance to anticancer therapies.136, 137
3.4 Adipocytes-derived exosomes
Recent studies suggest that adipocyte-produced extracellular vesicles (EVs) play crucial roles in obesity and related conditions.138 Exosomes are a type of membrane-encapsulated EVs that range in size from 30 to 100 nm. They are released into the extracellular microenvironment by a variety of cell types, including adipocytes and cancer cells, and are transported by different body fluids.139 The exchange of the exosome content, which consists of messenger RNAs (mRNAs), long non-coding RNAs (lncRNAs), microRNAs (miRNAs), transcription factors, proteins, and lipids,140, 141 mediates cell-to-cell biological communication and, in particular, the metabolic symbiosis between adipocytes and various types of cancer cells.142
According to a study by Clement et al.,143 adipocytes generate large amounts of exosomes that are intended to convey proteins essential in lipid metabolism, including FAO enzymes. These exosomes may play a role in tumor progression that spreads far from AT, such as the early stages of melanoma. The authors demonstrated that the quantity of adipocyte-derived exosomes released by adipocytes144 and their cargo145 are influenced by obesity or high-fat dietary interventions. Recent findings show that adipocyte-derived exosomes from patients affected by obesity are more abundant in proteins that facilitate cancer cell invasion and migration by raising FAO.142, 146 Taken together, these observations indicate that cancer patients with an obesity condition would likely benefit from FAO inhibitors and/or compounds that prevent EV uptake.
Moreover, a review by Bandini et al.147 discusses how miRNAs secreted by adipocytes are involved in breast cancer progression. They reported a study that demonstrated that miR-144 and miR-126 are carried at a high level by exosomes released after tumor cell-adipocyte interaction and that these miRNAs are responsible for the reprogramming of energy metabolism to facilitate tumor progression.148
Finally, Wang et al.149 demonstrated that exosomes from hepatocarcinoma cells may be actively integrated by adipocytes to transform them into tumor-promoting cells (exo-adipocytes). These were then proven to boost tumor development and promote angiogenesis in vivo. According to these findings, there is a complex crosstalk between adipocytes and cancer cells mediated by EVs through which cancer cells can alter AT to create a microenvironment that is favorable to tumor growth.149
3.5 Lipid peroxidation is essential for ferroptosis-mediated tumor suppression
Ferroptosis is a recently discovered sort of cell death implicated in various medical conditions and also in different types of cancer, such as large B-cell lymphoma (LBCL), renal cell carcinoma, liver cancer, cervical carcinoma, osteosarcoma, and prostate adenocarcinoma.150 Ferroptosis is a programmed necrosis, iron dependent, and ROS dependent, whose specific features clearly distinguish it from the other forms of cell death.151 It is primarily characterized by cytological changes, such as diminished or absent mitochondrial crests and destruction of the external mitochondrial membrane.151 Plasma membrane loss of selective permeability is triggered by cell abnormalities caused by massive lipid peroxidation that might be because of ROS accumulation.152, 153 The main driving factor behind ROS formation is the excessive presence of iron, which originated from abnormal iron metabolism.154 ROS play a variety of roles in cancer, including promotion of cell growth and survival, DNA damage, genetic instability, cell death induction, and increase of treatment resistance.155 To decrease ROS levels and keep redox equilibrium, tumor cells express significant quantities of antioxidant proteins. When this redox equilibrium is irreversibly disrupted, cancer cell death happens153 (Figure 3). As a result, ferroptosis is progressively recognized as an adaptive ability to eradicate cancerous cells. By eliminating cells that are lacking in essential nutrients or that are damaged by infection or environmental stress, ferroptosis plays a crucial role in the reduction of carcinogenesis.156

Because PUFAs are easily damaged by ROS, boosting their incorporation into membrane phospholipids is crucial to increase cancer cell susceptibility to ferroptosis. Lipid-peroxyl radicals produced in turn oxidize nearby PUFAs, triggering a chain reaction that, if without restraint, results in cell death.152 Magtanong et al.157 revealed that exogenous monounsaturated fatty acids (MUFAs) prevent ferroptosis by displacing PUFAs from membrane phospholipids. Considering these findings, it can be assumed that FA desaturation is closely related to processes that drive programmed cell death. Accordingly, cancer cells need to maintain the correct FA desaturation state to survive.158
Hence, lipid metabolism significantly affects the cells' sensitivity to ferroptosis. Evidence in breast cancer, fibrosarcoma, and prostate cancer cells supports the idea that cancer cells may protect themselves by simply depleting membrane PUFAs, given the high demand for PUFA–phospholipids in ferroptosis.157, 159, 160 To enable the reduction of the amount of PUFA-derived ROS in membranes and increase their resistance to lipid peroxidation, cancer cells promote de novo lipogenesis to produce more saturated FAs (SFAs) and MUFAs.13
Further research revealed that the molecule erastin could induce ferroptosis and, consequently, the death of cancer cells. Erastin prevents cancer cells from the uptake of cystine, weakening their antioxidant defenses and ultimately causing their death by ferroptosis.153 In addition, the combination of the ferroptosis-inducer erastin with chemotherapeutic treatments, such as cytarabine, cisplatin, doxorubicin, and temozolomide, resulted in a notable synergistic antitumor effect.154
Additionally, ferroptosis can attract and activate immune cells to the tumor sites by releasing chemoattractant molecules. This increases the chance that a ferroptosis-inducer could serve as a suitable enhancer for antitumor immunotherapy treatments like checkpoint inhibitors.161 Nevertheless, a study by Zhang et al.162 showed that a high body fat ratio was linked to cancer progression and a poor prognosis, partly because of a reduction in ferroptosis. They demonstrated that exosomes secreted by AT inhibit lipid ROS production, thus decreasing ferroptosis susceptibility and promoting chemotherapy resistance in colorectal carcinoma cells.
4 DIET AS A SOURCE OF EXOGENOUS LIPIDS
4.1 Pro- and anti-inflammatory dietary fatty acids
In cancer cells, lipids and FAs can be produced either by endogenous lipid synthesis, thanks to the mobilization of collected fat from adipocytes, or exogenously through diet. Some essential FAs, such as PUFAs, are not synthesized by the body and must be obtained through diet.163 A study by Hopperton et al.164 displayed that both exogenous and endogenous FAs are esterified to the same lipid and phospholipid classes and in the same proportions, suggesting that they are likewise important in stimulating cancer pathogenicity.
Previous studies have acknowledged diet and nutrition as partly responsible for cancer risk.165 Epidemiological, preclinical, and clinical research have related obesity and excess calorie consumption with heightened chances of tumor occurrence, while a low-carbohydrate, low-fat diet is linked to lower cancer incidence.24, 166 Numerous evidences indicated a direct link between high dietary fat intake and the risk of developing colorectal, liver, breast, pancreatic, gastrointestinal, and prostate cancers.167 This connection might result from a combination of increased consumption of some FAs that support cancer growth and/or decreased intake of other FAs that act as preventives for cancer progression.168 Indeed, FAs can have pro- or anti-inflammatory effects.169, 170 Saturated short-chain fatty acids (SCFAs), such as butyrate, propionate, and acetate, are produced mainly by microbial fermentation of plant-based fibers and have anti-inflammatory properties.171 These lead to cell cycle arrest and cause antiproliferative and proapoptotic effects in cancer cells, in particular in colon cancer.172
Another class of FAs are medium-chain fatty acids (MCFAs), which are contained in coconut oil and palm kernels and must be assembled into chylomicrons to be transported towards the liver, unlike the other FAs. The most common MCFAs include capric acid (C:10), lauric acid (C:12), capric acid (C:6), and caprylic acid (C:8). Similar to SCFAs, these MCFAs have been found to possess anticancer properties in in vitro experiments conducted on human breast tissue, skin, and colorectal cancer cells.173
However, saturated long-chain FAs (LCFAs), found in dairy fat, coconut oil, palm kernel oil, peanut oil, and meat lipids, have generally been linked to inflammatory effects and tumorigenesis, especially palmitic acid and stearic acid.174 While SCFAs and MCFAs can passively permeate the cell membrane, LCFAs need a fatty acid translocase, or CD36, for their uptake.175 In addition to this, a study by Daquinag et al.176 suggested that CD36 is also involved in LCFAs mobilization from AT, thus stimulating tumor growth and facilitating resistance to chemotherapy. CD36 is expressed in both adipocytes and endothelial cells, which are constituents of vessels in AT, and to a much lesser extent on the cancer cell surface. This led the authors to assume that the key mediator of LCFAs transfer from AT to tumors is exactly CD36 in the endothelium and adipocytes.176
Moreover, van Dijk et al.177 discovered a connection among AT inflammation, the presence of SFAs, and the absence of MUFAs. This is in line with a study that demonstrated that the expression of inflammatory genes in AT was enhanced by an SFA-rich diet, while it decreased during a MUFA-rich diet.178
Concerning the unsaturated FAs, docosahexaenoic (DHA) and eicosapentaenoic (EPA) acids, both ω-3 PUFAs which can be found in fish and other seafood, have anti-inflammatory actions and assist in repairing the damages of inflammation. They are related to a lower incidence of prostate,179 colorectal,180 and lung cancers.181 Conversely, arachidonic acid (AA), an ω-6 PUFA that can be found mainly in the fatty parts of meats and fish, is a precursor of eicosanoids that cause inflammation182 and is directly linked to a higher risk of colorectal cancer in both sexes.183 It has been shown that the rise of cancer risk, especially for breast, prostate,184, 185 and oral cancers, may also be linked with variations in the dietary ⍵-6/⍵-3 ratio.186
Exploring this important issue in patients is challenging given that a diet consists of many different foods.23 By examining the whole nutritional intake, researchers can understand the interplay between various foods, along with the influence on tumor risk of dietary elements associated with each other.187 However, the authors of the Lien et al. study26 found the deciphering of dietary data challenging because of the variability of the effects that a particular diet exerts on certain tumors. Considering these challenges, animal models have been used to unravel mechanisms and processes linking diets with specific tumor features. For example, mass spectrometry can be applied to understand how the tumor metabolomic profile is altered in animal models fed with a particular diet.26 Dietary components can impact the growth of tumors by altering cancer cell metabolism through modifications in the access to and use of nutrients by cancer cells. Alterations of diet composition lead to shifts in plasma metabolite levels, which in turn influence metabolite levels in the TME and eventually modify cancer cell metabolism and response to therapy.188
4.2 Diet implication in cancer growth and development
Many studies have been published on this topic. In particular, low-carbohydrate diets have been thought to inhibit the growth of several types of cancers189 (Table 2). Among these are caloric restriction (CR), which consists of chronic reduction by 30–40% of total calorie intake typically through carbohydrate limitation,190 and ketogenic diet (KD), which is a high-fat diet with a low carbohydrate intake (typically <50 g CHO/day). Several studies have demonstrated that CR can reduce tumor growth of diverse tumor types, including breast,191 lung,192 prostate,193 brain,194 pancreatic,195 and colorectal cancers.196 Lien et al.,197 studying both CR and KD, discovered that only CR prevents lung and pancreatic ductal adenocarcinoma mouse tumor allografts from growing. This means that additional processes help decrease progression of these tumors and lower the blood glucose and insulin levels. In particular, the authors observed that CR changed the nutrient availability, which led to lower lipid levels in plasma and tumors, while they did not find it in KD. In the same study, the authors found that a diet low in carbohydrates and high in fats and protein consistently increased the overall survival time of pancreatic ductal adenocarcinoma patients. This occurred if fats and proteins were plant-based instead of animal-based, considering that the latter contains more saturated fats.197 Instead, in a review by Zhu et al.,198 the authors reported certain benefits of the KD including improved efficacy of many anticancer drugs in neuroblastoma, glioma, prostate, lung, pancreatic, bladder, endometrial, and breast cancers, as well as in acute myeloid leukemia. KD may also have a tumor growth-limiting effect in glioblastoma, colon, and breast cancers. This occurs because of its anti-inflammatory effect and the antitumor mechanisms of ketone bodies produced, besides lowering blood glucose levels.199
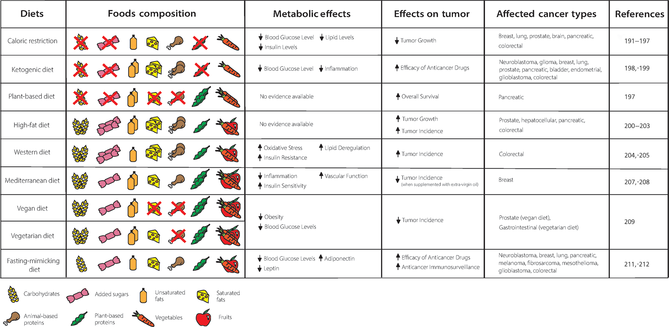 |
- Note: The table shows the food composition of the most popular diets, their known metabolic effects, their influence on tumor, and the affected cancer types. Double symbol: the food is highly consumed in the diet analyzed; crossed out symbol: the food is not consumed in the diet analyzed; (↑): increase; (↓) decrease.
Nevertheless, a high-fat diet (HFD), which, unlike KD, still contemplates considerable carbohydrate consumption, generally increases the incidence and growth of tumors, including colorectal,200 pancreatic,201 prostate,202 and hepatocellular carcinomas.203 Another similar diet, Western diet (WD), which is high in saturated fat and added sugars and low in fruits and vegetables, is linked to an increased risk of coronary artery disease (CAD), colon cancer, diseases with underlying oxidative stress, insulin resistance, AT inflammation, and lipid dysregulation, according to epidemiological studies.204, 205
Conversely, in massive general population cohorts, a diet with a high intake of fruits and vegetables has been correlated to a decreased risk of all-cause death and CAD events.206 As a result, many diets have been investigated for their ability to promote good health. One such diet is the Mediterranean diet, which emphasizes the consumption of whole grains, fruits, vegetables, legumes, seeds, and nuts. It also recommends the use of more olive oil and herbs as dish seasonings, and less butter and salt; it suggests to increase the consumption of fish and poultry and to decrease red meat. Additionally, it encourages people to eat more plant-based meals.207 In prospective studies, people with obesity who followed the Mediterranean diet saw improvements in circulating inflammatory markers, insulin sensitivity, and vascular function, which are metabolic syndrome indicators. These improvements lasted for up to 2 years.207 Significantly, a Mediterranean diet supplemented with extra-virgin olive oil in humans resulted in a lower incidence of breast cancer compared to the control group diet. This is probably because of the rich composition in polyphenols oleocanthal, oleuropein, hydroxytyrosol, and lignans of extra-virgin oil compared to other generic types of olive oil.208
Diets that are primarily plant-based, such as vegan and vegetarian diets, are associated with a lower risk of developing obesity, type 2 diabetes, ischemic heart disease, and some forms of cancer (gastrointestinal cancer for vegetarian diet and prostate cancer for vegan diet). A vegetarian diet involves avoiding the consumption of meat, poultry, fish, and their byproducts, while a vegan diet excludes all animal-derived products, including eggs, dairy products, and honey.209 These diets are characterized by a low-saturated fat intake and a high intake of fruits, vegetables, whole grains, legumes, soy products, nuts, and seeds (all rich in fiber and phytochemicals), which seem to improve serum lipid profile and blood sugar control.209 The reduction of red meat intake has been associated with favorable outcomes.210, 211 More specifically, the consumption of processed red meat was found to be associated with higher risk of colorectal cancer in two significant US cohorts.212
Finally, fasting-mimicking diets (FMDs) are specially formulated dietary plans that are extremely low in calories (300 to 1100 kcal per day on average), sugars, and proteins. They mimic many of the effects of water-only fasting with improved patient compliance and lower nutritional risk.213 According to Nencioni et al.,214 FMD cycles could be useful to enhance the effects of cancer therapies in glioblastoma, mesothelioma, melanoma, fibrosarcoma, neuroblastoma, lung, breast, pancreatic, and colorectal cancers, activating anticancer immunosurveillance, such as tumor-infiltrating lymphocytes, reducing levels of glucose and leptin and increasing levels of adiponectin.
From these results, it is clear that the journey to find a sure answer about the role of different diets in the development of many tumors is still long and challenging.
5 TARGETING LIPID METABOLISM AND ADIPOCYTES-BASED CELL THERAPY IN ONCOLOGY
5.1 Lipid metabolism-targeted agents
Obese CAAs can affect the efficacy of anticancer treatment by preventing tumor drug penetration, altering lipid-based metabolism, causing drug resistance in tumor cells, increasing abnormal vasculature, impairing immune cell function, and supporting tumor cell survival.215 Therefore, blocking these mechanisms is a novel promising strategy to develop innovative cancer therapies targeting the TME.216
In particular, the patients with obesity have been shown to exhibit a poor outcome after traditional treatments including radiation, chemotherapy, and surgery for cancer, because of a deficient wound healing process, necessary for the latter type of therapy. Studies on how high adiposity affects radiotherapy have shown that a higher BMI is strongly correlated with its reduced efficacy.217 However, obesity seemed to be associated with improved outcomes after treatment with immune checkpoint inhibitors. This has been called “the obesity paradox,” and the principal theory related to this phenomenon is the ability of AT to modify the phenotype of immune cells in a dysfunctional profile that is susceptible to treatment because of increased expression of the PD-1 target molecule.217 The metabolic environment linked to obesity triggers monocytes to transform into tumor-associated macrophages, leading to chemoresistance and unfavorable response to chemotherapy, which are further mediated by adipocytes acquiring stem-like features. These elements are important modulators of cell proliferation, neoangiogenesis, and inflammation, which favor cancer initiation and progression, as well as chemoresistance.218
One of the first solutions proposed to overcome complications correlated to obesity, was to combine well-known, affordable, and safe metabolic therapies with current cancer therapies to increase their efficacy, for instance, using metabolic drugs that currently treat widespread illnesses like type 2 diabetes219 and cardiovascular disease.220 These pathologies involve dysregulated lipid metabolism, and therapies that target these common pathways may offer chances to improve the effectiveness of cancer therapy. Proliferation of tumor cells would eventually be inhibited by depletion of lipids used for energy, and by boosting ROS levels, raising the sensitivity to additional stress and increasing therapeutic responsiveness.221
Small-molecule inhibitors, which can provide enzyme inhibition with molecular specificity, are another interesting family of drugs discovered in recent years. Acetyl-CoA carboxylase (ACC), in particular, modulates de novo FA synthesis222 and can be present in two isoforms (ACC1 and ACC2), which are encoded by the genes ACACA and ACACB, respectively. Small-molecule inhibitors of these two genes have been found and tested for their anticancer effects, with encouraging preclinical evidence. The ability of ND-646 to block both the ACACA and ACACB isoforms, thus stopping the growth of non-small cell lung cancer (NSCLC), was first reported in 2016.223, 224 Another small-molecule inhibitor, the liver-specific ND-654, was studied in a murine model of hepatocellular carcinoma (HCC), and it was shown to hinder liver lipid synthesis, reducing tumor formation.225 Both of these drugs were evaluated in combination with standard chemotherapy and demonstrated synergic inhibition by decreasing tumor size and incidence compared to monotherapy.223, 225
Other studies focused on trying to develop small-molecule inhibitors of stearoyl-CoA desaturase (SCD), a crucial enzyme involved in the production of MUFAs from their precursors, SFAs.226 According to recent evidence, ovarian cancer cells die by ferroptosis and apoptosis when SCD1 is blocked. This might be related to changes in the composition of membrane phospholipids brought on by the SCD1 inhibitors MF-438 and CAY10566.227
Another appealing therapeutic strategy is targeting lipid uptake.131 Recent evidence has shown that CD36 is a viable therapeutic target in cells that begin to metastasize.57 To mediate the specific transport of FAs into cells, CD36 needs co-transporters, which are often members of the FA transport protein (FATP)/SLC27A family.228 According to the study by Zhang et al.,131 FATPs are expressed by melanoma cells and are responsible for transporting lipids from adipocytes to tumor cells. Thus, melanoma cells co-expressing CD36 and FATPs may be more generally implicated in both tumor growth and invasion. This finding is supported by the discovery of a substantial positive connection between FATP1 expression and lipid content in tumor cells.131 Thrombospondin-1 is one of the ligands of CD36, and their bond can have anti-angiogenic function, contributing to cancer cell growth inhibition.229 Therefore, the anticancer properties of several thrombospondin analogs are now being investigated, including VT1021, which is undergoing phase I clinical trials for the treatment of several primary tumors (NCT03364400). Moreover, two distinct antibodies, FA6.152, which blocks all the activities of CD36, and JC61.3, which blocks only the uptake of FAs and LDLs, were both proved to be efficient and specific for tumors with lymph node metastases.57 Also targeting FABP4 has been demonstrated to be effective for the treatment of tumors metastasizing to visceral AT, such as ovarian, gastric, and colon cancers.50 FABP4 was found to be substantially expressed only in cancer cells at the adipocyte interface in a co-culture model.50 It is interesting to note that co-cultivating breast and colon cancer cells with adipocytes increased cancer cell production of FABP4. For this reason, it can be hypothesized that FABP4 inhibition would decrease FA uptake by cancer cells and slow tumor growth.50
A potential clinical approach is to prevent cancer cells from using FAs as a source of energy. In particular, it has been demonstrated in colon and breast cancer cells that etomoxir, an irreversible inhibitor of carnitine palmitoyltransferase-1 (CPT-1) in the mitochondria, potentiated the effect of the tested anticancer drugs by blocking the β-oxidation of FAs causing reduction of ATP levels, ROS generation, and apoptosis, as well as a collection of some cytotoxic LCFAs.230 However, because of serious adverse effects, etomoxir clinical use is currently restricted.231 Reversible CPT-1 inhibitors offer a different option along with fewer negative side effects. ST1326, a reversible CPT-1 inhibitor, has shown anticancer properties in numerous non-solid tumors.232, 233 Moreover, there is pre-clinical evidence for the use of additional inhibitors of mitochondrial FAO, such as perhexiline (another CPT-1 inhibitor) and trimetazidine (an inhibitor of mitochondrial long-chain 3-ketoacyl coenzyme A thiolase [LCTH], which catalyzes the last step in mitochondrial β-oxidation). However, these molecules are currently in registered clinical trials for cardiac pathologies and type 2 diabetes only.221
As most cancers increase FA production, blocking this mechanism is a straightforward strategy to decrease the presence of FAs in cancer cells. Fatty acid synthase (FASN), a lipogenic gene, is often overexpressed in cancer cells and is a potential target for therapy.234, 235 Many studies have shown that FAS activity inhibition leads to apoptosis in cancer cells. The FAS action could be blocked by different therapeutic substances. For example, cerulenin permanently alters the target enzyme function236 although it is not selective for FAS.237 The first synthetic FAS-I inhibitor with notable anticancer action is C75.234 However, C75 causes significant weight loss in animals, and it induces anorexia in proportion to the dose increase. Lately, C93 has been characterized as a more specific FAS inhibitor.236 Orlistat is an oral medication that inhibits the thioesterase domain of FAS and is used for managing obesity.236 It can induce apoptosis in various cancer cell lines, but this does not occur in not normal cells.236
Finally, TVB-3166 and TVB-3664 are well-tolerated molecules that selectively inhibit FASN. This, in turn, suppresses the PI3K-Akt-mTOR pathway and β-catenin signaling to hinder cancer cell growth and promote their death.238
5.2 Adipocyte-based cell therapy
In recent years, adipocytes have been effectively used as vectors for anticancer drugs. The adipocyte-based delivery system shares features with other cell-based systems, including high biocompatibility, prolonged circulation in vivo after systemic injection, and strong affinity towards tumors and inflammatory areas. Furthermore, hydrophobic medicines can be effectively encapsulated because of the presence of lipids in adipocytes. Lipid droplets can gather during the lipolysis process triggered by tumor cells to support their metabolism, and this can be used as a “Trojan horse” method to provide local and long-lasting delivery of various drugs at the tumor sites.239 As an example, Wen et al.240 engineered adipocytes to act as a reservoir to transport anticancer drugs at the tumor sites by taking advantage of lipid metabolism. They discovered that adipocytes loaded with a ROS-responsive doxorubicin prodrug (pDOX) and rumenic acid (RA) might negatively affect tumor cells by activating the lipid metabolic pathway controlled by FABP4. Likewise, Aoki et al.241 created a drug delivery system (DDS) based on ASCs that carried poly lactic-co-glycolic acid (PLGA), conjugated with pirarubicin. Pirarubicin has an inhibitory activity on DNA replication and can release anti-pancreatic cancer factors. Thanks to the infiltration of ASCs into the tumor site and to the dual effect of released pirarubicin, pir-PLGA nanoparticles (NPs) encapsulated in ASCs were able to inhibit the growth of pancreatic tumors, causing cancer cell apoptosis. In addition, when compared to pirarubicin treatment alone, pir-PLGA NP-loaded ASCs inhibited tumor development at very low doses with few side effects.
6 CONCLUSIONS AND FUTURE PERSPECTIVES
Adipocytes and AT have been recognized to play a crucial role in tumor development and progression. Adipocytes close to or distant from tumors establish a vicious cycle with cancer cells through adipokines, exosomes, lipids, and fatty acids exchange, promoting tumor growth, progression, metastasis, and resistance to therapy. Adipokines can act by stimulating pathways involved in cancer cell adhesion, migration, invasion, angiogenesis, and inflammation. FAs and lipids supply the cells growing need for metabolic substrates in support of proliferation and function as mediators or signaling molecules. Cancer cells, in turn, can reprogram adipocytes towards a more inflammatory state stimulating the release of lipids and the secretion of adipokines and chemokines. These mechanisms are enhanced in obesity, which is associated with an increased risk of developing certain cancers; this is also probably because of the intense use of lipids by tumor cells. For all these reasons, increasing effort has been addressed to understand mechanisms that lead to obesity, and novel therapeutic strategies based on the targeting of cancer metabolism are under preclinical and clinical development. The direction taken is promising but we must consider that responses can be highly variable, depending on the tumor types. The mapping of specific vulnerabilities to the different metabolic inhibitors for each tumor type, as well as the identification of potential synergistic effects with standard therapies, is required.
Diet is crucial in this context, providing the correct balance of lipids, which can have pro- or anti-inflammatory effects on the human body. However, nutritional interventions for cancer patients and close monitoring by nutritionists throughout treatment have not been fully established. Understanding the mechanisms by which diet modifies the cancer cell metabolism and tumor features is challenging, considering the diet complexity. Therefore, we believe a prospective serum metabolome profiling of cancer patients under specific diet regimens would add important information. Finally, it is necessary to study the correlation between the metabolome of tumors and biological, molecular disease features, as well as patient clinical outcomes, including treatment response.
AUTHOR CONTRIBUTIONS
Chiara Calabrese and Chiara Liverani conceived, wrote, and edited the manuscript. Giacomo Miserocchi, Alessandro De Vita, Chiara Spadazzi, Claudia Cocchi, Silvia Vanni, Sofia Gabellone, Giovanni Martinelli, Nicoletta Ranallo, and Alberto Bongiovanni wrote the manuscript. Giacomo Miserocchi conceived and drafted the figures. All authors reviewed and approved the manuscript.
ACKNOWLEDGEMENTS
This work was supported by an Italian Ministry of Health Grant for the project GR-2019-12370014. The authors thank Marcia Jacqueline Myrie and Cristiano Verna for their editorial assistance. Open access funding provided by BIBLIOSAN.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest.
Open Research
DATA AVAILABILITY STATEMENT
The datasets generated and/or analyzed during the current study are available from the corresponding author upon reasonable request.

