

On the path to extinction: Inbreeding and admixture in a declining grey wolf population
Abstract
Allee effects reduce the viability of small populations in many different ways, which act synergistically to lead populations towards extinction vortexes. The Sierra Morena wolf population, isolated in the south of the Iberian Peninsula and composed of just one or few packs for decades, represents a good example of how diverse threats act additively in very small populations. We sequenced the genome of one of the last wolves identified (and road-killed) in Sierra Morena and that of another wolf in the Iberian Wolf Captive Breeding Program and compared them with other wolf and dog genomes from around the world (including two previously published genome sequences from northern Iberian wolves). The results showed relatively low overall genetic diversity in Iberian wolves, but diverse population histories including past introgression of dog genes. The Sierra Morena wolf had an extraordinarily high level of inbreeding and long runs of homozygosity, resulting from the long isolation. In addition, about one-third of the genome was of dog origin. Despite the introgression of dog genes, heterozygosity remained low because of continued inbreeding after several hybridization events. The results thus illustrate the case of a small and isolated wolf population where the low population density may have favoured hybridization and introgression of dog alleles, but continued inbreeding may have resulted in large chromosomal fragments of wolf origin completely disappearing from the population, and being replaced by chromosomal fragments of dog origin. The latest population surveys suggest that this population may have gone extinct.
1 INTRODUCTION
Human encroachment has led to historic declines in most large carnivores over the last century through a combination of habitat modification and direct persecution (Ripple et al., 2014). Smaller numbers of individuals can result in decreased density, reduced distribution range and increased fragmentation of remaining populations. As population size decreases, stochastic demographic and environmental factors start to determine the dynamics of the populations, random genetic drift leads to the loss of potentially beneficial genetic variants, the power of natural selection is diminished, and inbreeding (and potential for inbreeding depression) increases. All these are expressions of Allee effects, the decrease in individual fitness as the population size falls (Courchamp, Clutton-Brock, & Grenfell, 1999; Kramer, Dennis, Liebhold, & Drake, 2009). Another Allee effect is the decrease in social interactions which will further increase the level of threat and risk of extinction for a population because it becomes harder to find a mate, social groups become smaller resulting in decreased opportunities for cooperative hunting or defence, etc. While fragmentation into small populations is likely to negatively affect all vertebrates, carnivores and social animals are particularly susceptible. Carnivores are typically found at low densities, and most small- and medium-sized habitat patches are insufficient to maintain viable populations. Similarly, social species require larger ranges and higher numbers of individuals per reproductive unit than solitary species (although sociality may contribute to buffer Allee effects, see Angulo, Rasmussen, Macdonald, & Courchamp, 2013). In this sense, allee effects have been hypothesized to play an important role in the decline of African wild dog (Lycaon pictus) populations (Courchamp, Clutton-Brock, & Grenfell, 2006).
A good example of a social carnivore often threatened by reduced population size and fragmentation is the grey wolf (Canis lupus L.). The grey wolf was historically distributed over the entire Holarctic, but direct persecution and habitat and prey loss have led to a reduction in distribution and density (Boitani, 2003). However, a series of small populations survived and some of them have increased in size and expanded in the last decades (Chapron et al., 2014). Because the grey wolf is a top predator and a very charismatic species, some of these small and fragmented populations have received the attention of researchers for many years. Thus, it has been possible to monitor the expansion of the Scandinavian wolf population from just three founders (Åkesson et al., 2016; Liberg et al., 2005; Vilà, Sundqvist, et al., 2003; Vilà, Walker, et al., 2003), the long-term survival of the Isle Royale wolves from a similarly reduced founding event (Peterson, Thomas, Thurber, Vucetich, & Waite, 1998), and the sustained survival in captivity of the Mexican wolves despite their disappearance in the wild (Hedrick & Fredrickson, 2008; Hedrick, Miller, Geffen, & Wayne, 1997). These cases led some to believe that wolves were almost immune to the deleterious effects of inbreeding depression and that small, highly inbred populations could be perfectly viable. However, the evidence accumulated during recent years clearly shows that this is not the case. These populations of wolves are becoming examples of the burden that deleterious alleles can impose on natural populations as a result of inbreeding. For example, skeletal abnormalities have been observed in Scandinavian and Isle Royal wolves (Räikkönen, Bignert, Mortensen, & Fernholm, 2006; Räikkönen, Vucetich, Peterson, & Nelson, 2009), reduced reproductive output in Scandinavia (Liberg et al., 2005), reduced sperm quality (Asa et al., 2007) and fitness declines (Fredrickson, Siminski, Woolf, & Hedrick, 2007; Hedrick, Peterson, Vucetich, Adams, & Vucetich, 2014) in Mexican wolves. These studies share a unique characteristic that has made them excellent models to understand the consequences of Allee effects associated with population decline and fragmentation: The populations have been continuously monitored in the field and in the laboratory for a long time period. However, this is an exceptional situation and in most cases the knowledge available about the demographic history and loss of genetic diversity is nonexistent for critically threatened wildlife populations. One of these poorly known but critically threatened populations is the Spanish Sierra Morena wolf population.
The northwest of the Iberian Peninsula, which includes Spain and Portugal, houses a moderately large population of wolves—currently about 2,200–2,500 individuals (Hindrikson et al., 2017)—that has remained isolated from other European wolf populations since the mid-19th century. This is the largest population in Western Europe. However, hundreds of kilometres south of the southern limit of the distribution of this wolf population lays Sierra Morena, a mountain system where a very small wolf population has lived in complete isolation for perhaps as much as half a century (Blanco & Cortés, 2007; Supporting Information Figure S1). This population is separated from the northern wolf population by extensive agricultural areas, large rivers and highly developed areas. Different reports over the last 25 years have indicated that this population is either composed of a few groups of wolves, one or none. An accurate knowledge about the population has been impossible because the wolf range is located on private hunting estates with very limited and difficult access. The last robust evidence of wolves in the region derives from faeces identified as wolf using mitochondrial DNA in 2012 and 2013, and one breeding group was located in 2013 (Junta de Andalucía, 2015) but the latest Spanish National Census, from 2016, considers that there is not a breeding population (MAGRAMA, 2016). However, logistic difficulties prevent repeated, reliable surveys. The assessment of the consequences of fragmentation and isolation on the status and health of this population has been unfeasible.
Fortunately, genomic approaches can help overcome some of the limitations inherent in studying this mysterious population. Instead of requiring samples from an important fraction of the population to monitor it over time, genome sequences of potentially representative individuals could inform about population-wide losses of diversity, admixture between divergent lineages or demographic history. In this study, we use the complete genome sequence of a single wolf from the Sierra Morena population that was road-killed in 2003 and compare it to whole-genome sequences obtained from three northern Spanish wolves and other previously published dog and wolf genome sequences. The comparison of these sequences can help us to better understand the long-term survival of this population and the consequences of remaining at a small size for multiple generations.
2 MATERIALS AND METHODS
2.1 Sampling and sequencing
We generated the whole-genome sequences of two Iberian wolves: one from a captive breeding population belonging to the European Endangered Species Programmes (EEP, henceforth labelled wEEP) and one from southern Spain (Sierra Morena, wSierraMorena). The captive wolf sample consisted of whole blood drawn during a routine veterinary examination from a captive-born wolf at the Zoological Park of Barcelona that descends from the Iberian northwestern population and the sequence obtained for this individual was used in parallel in another study (Botigué et al., 2017). Previous genetic analyses revealed that most of the diversity present in the wild wolf population was also present in the captive population due to the large number of founders used (Ramirez et al., 2006). The Sierra Morena wolf sample comes from an animal found road-killed in 2003 in northeastern Andalusia (southern Spain), with general wolf-like appearance that did not suggest dog admixture and preserved by the Andalusian Regional Government. Illumina libraries were constructed following manufacturer's instructions (Supporting Information Appendix S1) and sequenced in the CNAG (Centre Nacional d'Anàlisi Genòmica, Barcelona, Spain). These whole-genome sequences were compared to the previously published genome sequences of 11 dogs (from 11 breeds) and 14 wolves from 10 populations from across the distribution range and including two more Iberian wolves (from the northwestern population and sampled in Spain and Portugal, wSpain and wPortugal; Fan et al., 2016; Freedman et al., 2014; Serres-Armero et al., 2017; Wang et al., 2013).
2.2 Mapping, SNP calling and filtering
All the sequences were mapped to the dog reference genome (canFam3.1) using bwa version 0.6.1 (Li & Durbin, 2009) with the quality trimming parameter set to a Sanger quality score of 15 and default parameters. Next, we used picard tools version 1.70 (http://broadinstitute.github.io/picard/) to remove PCR duplicates and gatk version 2.5 (McKenna et al., 2010) to perform indel realignment. The resulting files were used for variant calling. We produced a preliminary set of 19,640,837 SNPs using gatk's UnifiedGenotyper and VariantFiltration with the recommended filtering parameters for the case in which Variant Quality Score Recalibration (VQSR) is not available (Van der Auwera et al., 2013). To avoid low complexity regions and gaps (Li, 2014), mappable regions were obtained using gem mappability (Derrien et al., 2012) and custom Perl scripts. We obtained a final data set containing 18,956,547 SNPs that were identified in all genome sequences. The DepthOfCoverage tool implemented in gatk was used to calculate average depth of coverage of this final set.
2.3 Diversity analysis and inbreeding
To explore the genomewide distribution of genetic variability in the Iberian wolf samples, we looked at heterozygosity (heterozygous positions per callable base) across the genome in 1 Mb windows (with 200 Kb of overlap). Only windows with a 100-Kb minimum callable region were considered and, to avoid coverage differences between samples, we removed variants in noncallable sample-specific regions with gatk's CallableLoci (minimum base quality of 20 and a depth range in the mean ± 5 of the autosomal read depth based on the individual coverage, Table 1).
| Sample | Species | Population | Cov | Het (×1000) | FROH | Dog blocks |
|---|---|---|---|---|---|---|
| wSierraMorena | Grey Wolf (Iberia) | South Spain | 43.94 | 1.093 | 0.42 | 31.88 |
| wSpain | Grey Wolf (Iberia) | NW Iberia | 22.68 | 1.543 | 0.15 | 14.30 |
| wPortugal | Grey Wolf (Iberia) | NW Iberia | 24.30 | 1.183 | 0.30 | 2.94 |
| wEEP | Grey Wolf (Iberia) | NW Iberia | 22.74 | 1.466 | 0.15 | 3.20 |
| Wolf Croatia | Grey Wolf (Eurasia) | Croatia | 6.98 | 1.473 | 0.09 | |
| Wolf China | Grey Wolf (Eurasia) | China | 26.36 | 1.484 | 0.23 | |
| Wolf India | Grey Wolf (Eurasia) | India | 24.90 | 1.814 | 0.01 | |
| Wolf Iran | Grey Wolf (Eurasia) | Iran | 26.27 | 1.781 | 0.03 | |
| Wolf Israel | Grey Wolf (Eurasia) | Israel | 6.01 | 1.507 | 0.05 | |
| Wolf Italy | Grey Wolf (Eurasia) | Italy | 5.81 | 0.321 | 0.51 | |
| Wolf Great Lakes | Grey Wolf (America) | North America | 24.34 | 1.831 | 0.08 | |
| Wolf Yellowstone A | Grey Wolf (America) | Wyoming | 25.73 | 1.546 | 0.18 | |
| Wolf Yellowstone B | Grey Wolf (America) | Wyoming | 24.07 | 1.586 | 0.13 | |
| Wolf Yellowstone C | Grey Wolf (America) | Wyoming | 5.41 | 1.485 | 0.09 | |
| Wolf Mexico A | Grey Wolf (America) | Mexico | 23.59 | 0.038 | 0.70 | |
| Wolf Mexico B | Grey Wolf (America) | Mexico | 5.23 | 0.120 | 0.70 | |
| Airedale Terrier | Dog | 7.33 | 0.644 | 0.44 | ||
| Basenji | Dog | 12.35 | 0.686 | 0.34 | ||
| Boxer | Dog | 29.33 | 0.664 | 0.41 | ||
| Chinese Crested | Dog | 19.17 | 0.763 | 0.41 | ||
| Chinook | Dog | 7.84 | 0.807 | 0.39 | ||
| English Cocker Spaniel | Dog | 9.66 | 1.044 | 0.25 | ||
| Kerry Blue Terrier | Dog | 15.83 | 0.688 | 0.44 | ||
| Labrador Retriever | Dog | 10.80 | 1.105 | 0.20 | ||
| Miniature Schnauzer | Dog | 5.47 | 0.767 | 0.32 | ||
| Soft Coated Wheaten Terrier | Dog | 17.18 | 0.703 | 0.41 | ||
| Standard Poodle | Dog | 12.63 | 1.016 | 0.28 |
Notes
- Sample name, species and population of origin for each sample whose genome sequence has been studied here.
- Cov: sequencing coverage; Het (×1,000): heterozygosity (heterozygous position per 1,000 bp); FROH: inbreeding coefficient; Dog blocks: percentage of dog ancestry blocks across the genome. wSierraMorena and wEEP were sequenced for this study, the others are from the literature (Freedman et al., 2014).
Runs of homozygosity (ROHs) may reflect historical population demographics or homozygosity by descent (Li et al., 2006). Long ROHs (>1 Mb) are indicative of autozygosity, inbreeding or admixture (Boyko et al., 2010; Pilot et al., 2014). Due to this association with recent past demography, we conservatively considered ROHs when at least two consecutive nonoverlapping 1 Mb windows (≥2 Mb) fell under a heterozygosity threshold of 0.0005 (Supporting Information Figures S2 and S3). To calculate the inbreeding coefficient based in runs of homozygosity (FROH), we applied the modified definition of Keller, Visscher, and Goddard (2011):
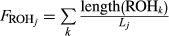
where ROHk and Lj are the kth ROH and the individual j's callable genome length. We used a different callable genome length for each sample based on its mappability.
2.4 Ancestry analysis
To assess the ancestry of each Iberian individual, we performed an admixture (Alexander, Novembre, & Lange, 2009) analysis, which uses the same likelihood model as structure (Falush, Stephens, & Pritchard, 2007; Pritchard, Stephens, & Donnelly, 2000), and a principal component analysis (PCA) using the smartpca program implemented in the eigensoft package (Price et al., 2006). We reduced the number of variants by removing nonbiallelic and missing markers, filtering out with MAF < 0.01 and LD-pruned using plink version 1.07 (Purcell et al., 2007), with sliding-window size of 50 SNPs (with an overlap of 10 SNPs) and r2 = 0.5. The final data set used for the admixture and PCAs contained 4,558,774 SNPs.
We further evaluated the ancestry for the Iberian wolves by comparing our data to previously published 48K SNP data sets including a large number of dogs and wolves from around the world (Boyko et al., 2010; vonHoldt et al., 2010, 2011). These data came from the Affymetrix Canine version 2 genomewide SNP mapping array, which uses CanFam2 assembly coordinates. To identify the same SNPs in our data sets, each of our whole-genome data was mapped and SNPs were called again to this assembly. After joining data sets, filtering by MAF and LD-pruned with plink as above, we obtained a set of 43,497 SNPs (43K data set). With this data set including more individuals, we repeated the admixture and PCAs. To assess the variability in the estimates obtained with admixture, the program was run five times for the genomic data set and three times for the 43K data set, with the number of groups (K) varying between 2 and 10, and a fivefold cross-validation (Alexander & Lange, 2011).
2.5 Introgression analysis
To assess the admixture between Iberian wolves and dogs, we identified alleles shared pairwise between each Iberian wolf and all other genomes (calculated over the entire SNP data set: not-pruned—15,807,997 SNPs—without nonbiallelic and missing markers). To determine regions introgressed from dogs in Iberian wolves, we used pcadmix version 1.0 (Brisbin et al., 2012) with a 50 SNP window size. This program estimates local ancestry via principal components analysis. As this program needs phased genotypes, the complete pruned data set was phased using shapeit version 2.644 (Delaneau, Zagury, & Marchini, 2012).To detect blocks of ancestry for the Iberian wolves, pcadmix was run with the 11 dogs as one ancestral population and six Eurasian wolves as the second. Three kinds of blocks were identified according to the origin of the chromosomal fragments: Dog/Dog, Wolf/Dog and Wolf/Wolf.
As most of the studies about hybridization in wolves have been carried out using a small panel of autosomal microsatellites, a microsatellite analysis was carried out with the Iberian wolf samples that apparently had hybrid origin (see Results). Our goal was to evaluate whether those reduced panels can identify different levels of admixed ancestry. We genotyped 10 autosomal microsatellite markers following the methods of Sastre et al. (2011). The resulting genotypes were compared to a previously published data set containing genotypes of 31 Iberian wolves and 32 dogs (Sastre et al., 2011). To assess the proportion of the genome that could come from dogs according to this more limited microsatellite data set, we carried out a Bayesian model-based clustering approach implemented in structure version 2.0 (Falush et al., 2007), running 100,000 Markov chain Monte Carlo repetitions and a burn-in of 10,000 iterations for K = 2.
3 RESULTS
3.1 Heterozygosity and inbreeding
The two Iberian wolf samples sequenced for this study, wSierraMorena and wEPP, were sequenced at high coverage (44× and 23×, respectively) to ensure reliable estimates of heterozygosity. The coverage for the other two previously sequenced Iberian wolves was also high (23× and 24× for wSpain and wPortugal, respectively), thus preventing deceptively low estimates for this isolated population (Table 1). Among these wolves, wSierraMorena had the lowest mean heterozygosity (1.09 × 10−3 heterozygous positions per bp, het/bp). wPortugal showed slightly higher heterozygosity (1.18 × 10−3 het/bp) while the other Iberian wolves had values close to 1.5 × 10−3 (Supporting Information Figure S2). The mean heterozygosity observed in the genome sequences of other Eurasian wolves was about 1.6 × 10−3 het/bp (except for the Italian wolf, originating from a highly inbred population, where it is lowest, 0.3 × 10−3; Table 1, Supporting Information Figure S3b), consistent with other genomewide studies (Freedman et al., 2014; Lindblad-Toh et al., 2005). American wolves also had similarly high values, except the highly inbred Mexican wolf (the samples studied here correspond to the Ghost Ranch lineage, a particularly inbred line; Supporting Information Figure S3b). On the other hand, purebred dogs had reduced heterozygosity (0.88 × 10−3 het/bp on average; Table 1, Supporting Information Figure S3a), as expected from their relative isolation and small effective population size (Calboli, Sampson, Fretwell, & Balding, 2008), but varied across breeds. The heterozygosity of wSierraMorena was similar to that observed in some of the purebred dogs and only lower than the most inbred wolf populations (Table 1, Supporting Information Figure S3).
Runs of homozygosity appeared in all Iberian wolves (Supporting Information Figure S4), but were much more frequent in wSierraMorena, which had chromosomes almost entirely homozygous (chromosomes 18, 27, 35, 37 and 38; Figure 1). This individual showed the largest ROHs at 40–60 Mb, and the inverse cumulative curve was clearly above all other Iberian wolves (Supporting Information Figure S4). The presence of long ROHs in wSierraMorena (Supporting Information Figure S4) implies high inbreeding few generations ago (see Thompson, 2013)). Although wPortugal also had some runs longer than 40 Mb, their distribution was similar to other Iberian wolves. wSpain and wEEP showed practically identical ROH cumulative curves, and almost all ROHs were shorter than 30 Mbp. The inbreeding coefficient showed that wSierraMorena was the most inbred Iberian wolf (FROH = 0.42). The least inbred Iberian wolves came from northern Spain, wSpain and wEEP (FROH = 0.15), and the inbreeding coefficient was intermediate for wPortugal (FROH = 0.30; Table 1). For other wolves, FROH tended to be much lower than for the Iberian wolves, with most values between 0.01 and 0.09 (Table 1). The exceptions to this pattern were the Italian (FROH = 0.51) and Mexican wolves (a captive population, FROH = 0.70), where high inbreeding has already been shown in previous studies and is in agreement with the expectations from their demographic history (Fabbri et al., 2007; Hedrick et al., 1997; vonHoldt et al., 2010, 2011). This value is also somewhat high for the Chinese wolf (FROH = 0.23), but the reasons for this high value are not clear. For the Yellowstone wolf population, founded recently from a relatively small number of founders, FROH varies between 0.09 and 0.18. As expected, purebred dogs have high inbreeding, with FROH in the range between 0.20 and 0.44. However, these values are similar or lower than the inbreeding estimated for wSierraMorena.

3.2 Introgression of dog alleles
A PCA using genomewide data (Figure 2a) showed that dogs and wolves separated along the first principal component (PC1, Figure 2a), while the second axis separated American and Eurasian wolves. Interestingly, wSierraMorena appeared intermediate between dogs and wolves and wSpain appeared somewhat separated from the other Iberian wolves. A PCA with a reduced representation of about 43K SNPs but including many more individuals from previous studies (Boyko et al., 2010; vonHoldt et al., 2010, 2011) revealed that Italian wolves were clearly separated from the other Eurasian wolves and showed that while wPortugal and wEPP clustered with other Iberian wolves, wSpain appeared somewhat separated and wSierraMorena was located segregated from all other wolves and shifted towards dogs along PC1 (Figure 2b). Subsequent axes on the 43K data set differentiated additional wolf populations (Supporting Information Figure S5).

Cross-validation error for the admixture analyses for the 4.5 million SNP data set suggested that the data were compatible with two clusters, K = 2 (Supporting Information Figure S6). At this level, dogs and wolves appeared clearly separated (Figure 3), but three wolves seemed to suggest introgression of dog alleles into wolf populations, two Iberian wolves (wSierraMorena and wSpain, with 31.5% and 10.4% of dog genome, respectively), and the sample from Israel. However, at higher K values only wSierraMorena consistently showed evidence of dog introgression. These higher K values also differentiated other wolf populations. Using the 43K data set, which included many more individuals, the cross-validation suggested that the data were best explained by a large number of clusters, at least K = 9 (Supporting Information Figure S6), but the exact number was not properly investigated because we only explored K = 2–10. In any case, the results reflect the fragmentation into distinct wolf populations and dog breeds that are likely to be completely isolated today. As before, at K = 2 this analysis also separated dogs and wolves and also suggested a slightly higher introgression of dog alleles into wSierraMorena (36.9%) and wSpain (17.7%; Supporting Information Figure S7). For the other two Iberian wolves, the putative introgression seems negligible (for wPortugal, 0.00% and 4.47% in the analyses with the genomic and 43K data sets; for wEEP, 0.00% and 3.28%, respectively). The 43K SNP analyses seemed to suggest higher rates of introgression, but this could be partly due to ascertainment bias in the microarray design, which maximizes dog variability (Boyko et al., 2010; vonHoldt et al., 2010, 2011).

The introgression of dog genes into the genome of these Iberian wolves came as a surprise, especially for wSpain, which in previous studies (Fan et al., 2016; Freedman et al., 2014) had been taken as a representative of pure northern Iberian wolves considering the low frequency of wolf–dog hybridization in this population as estimated in previous microsatellite analyses (Godinho et al., 2011; Pacheco et al., 2017). To assess to what degree a small panel of microsatellite markers could identify these admixed wolves, we typed them for 10 autosomal microsatellites previously used to separate dogs and wolves (Sastre et al., 2011). The estimate of dog ancestry obtained with this approach was 42.4% for wSierraMorena and only 0.6% for wSpain, with the probability interval not excluding the possibility of no introgression in this last sample (0% of dog genome; Supporting Information Figure S8). These results are very different from those obtained with genomic data for wSpain, which was not identified as admixed suggesting poor power to estimate ancestry due to the small number of markers.
The identification of 50-SNP blocks in the genome of Iberian wolves that could have originated in dogs revealed that almost a third of wSierraMorena's genome (31.9% of 50 SNP windows) came from dogs (Table 1, Figure 4). The proportion was 14.3% for wSpain. Interestingly, the distribution of blocks of dog ancestry in the two samples was very different. While wSpain showed a very large number of small blocks, usually pairing with blocks of wolf ancestry (Wolf/Dog blocks), wSierraMorena showed several regions with very large blocks of dog origin on the two homologous chromosomes (Dog/Dog blocks; for example, on chromosomes 2, 3, 4, 9, 17, 36 and 38; Figure 4). The other two Iberian wolves, wPortugal and wEEP, had only about 3% of putative blocks of dog ancestry, close to the proportion of dog ancestry suggested by previous analyses.

After removing the SNP windows that represented dog haplotypes in at least one of the admixed Iberian samples, a PCA showed the same groups of samples as before, but now all four Iberian wolf samples clustered in a group (Figure 2c) supporting the notion that the individuals that appeared separated in the previous analyses (Figure 2a) did so because of introgression of dog alleles. Still, while the three samples originating from the northwestern population practically shared the same position, wSierraMorena—originating from a small population putatively isolated for decades—appeared somewhat separated.
Consistent with this observation, the proportion of shared alleles for any of the Iberian wolves was highest when compared to other Iberian wolves, but somewhat lower for wSierraMorena (Supporting Information Figure S9a). The proportion of shared alleles was just slightly lower when comparing with a Central European wolf from Croatia, lower when comparing with Italian and Middle Eastern/Asian wolves, and the lowest, as expected, for American wolves. The proportion of alleles shared with dogs for wPortugal and wEPP was as low as the proportion shared with American wolves, suggesting that this sharing is result of a common evolutionary history more than admixture. The proportion of alleles shared with dogs was slightly higher for wSpain. On the other hand, for wSierraMorena the proportion shared with dogs was much higher and similar to the proportion shared other European wolf populations. From the alleles present in Iberian wolves (22,959,835 of 31,616,032 in the data set), wSierraMorena had a higher proportion of singletons (5%) and shared slightly more alleles with wSpain than with the other samples (Supporting Information Figure S9b). These results are consistent with extensive introgression of dog alleles into the genome of wSierraMorena and also a lower level of introgression into the genome of wSpain.
At the chromosomal level, separating blocks of wolf ancestry (Wolf/Wolf) from those containing dog haplotypes (Dog/Dog and Wolf/Dog) revealed that for wSpain (and the other wolves from northern Spain), blocks containing dog haplotypes tended to have high heterozygosity (Figure 5). On the contrary, heterozygosity for these blocks was lower than Wolf/Wolf blocks for wSierraMorena, many of them below the homozygosity threshold. While Dog/Dog blocks represented a very small portion of the chromosomes of wSpain and these blocks tended to have high heterozygosity (Figure 5), they added to more than 50% of the total length of some chromosomes in wSierraMorena (Figure 4) and in these cases the heterozygosity was remarkably low. This could indicate that the larger dog blocks derived from one single interbreeding event and that a small number of dog chromosomes have recently spread across the Sierra Morena wolf population. Subsequent inbreeding has led to identity by descent of many of these chromosomal fragments.

4 DISCUSSION
4.1 Iberian wolf populations
The whole-genome sequence of four Iberian wolves revealed that, after excluding regions affected by the introgression of dog alleles, the three northern wolves had a very similar genetic composition, clustering in one tight group in the PCA, while the wolf originated in Sierra Morena appeared somewhat differentiated (Figure 2c). This was expected due to the relative isolation of this southern population and probable intense drift. In addition, our results also showed an important level of inbreeding even in the most outbred individuals (FROH = 0.15 for wSpain and wEPP), suggesting inbreeding in deep history. This contrasts with the expectations, because the Iberian wolf population is the largest in Western Europe (Chapron et al., 2014) and its size is not known to have been dramatically smaller than today in the past. However, the result is consistent with a reduced effective population size previously reported based on microsatellite analyses (Sastre et al., 2011). This suggests that the size of the Iberian wolf population in the past may have been smaller than commonly assumed for the 20th century.
Our results also highlight the diversity of evolutionary patterns for the northern Iberian wolves. On the one hand, wPortugal revealed a surprisingly low heterozygosity and high inbreeding, comparable to that of full siblings (Table 1). Excluding the Sierra Morena wolf, these values were more extreme than for any of the other wolves included in the study except for the Italian and Mexican wolves, two highly inbred populations (Fabbri et al., 2007; Hedrick & Fredrickson, 2008; Hedrick et al., 1997; Lucchini, Galov, & Randi, 2004). The other nonadmixed northern wolf (wEPP) had diversity values more similar to other Eurasian wolves (Table 1). The Wolf/Wolf portions of the genome of wSpain had intermediate heterozygosity (Figure 5). The large difference in heterozygosity between these three wolves is surprising because the entire northern range for the Iberian wolves is just about 90,000 km2, and the linear distance between the sampling locations for the different individuals was under 250 km and apparently without intervening barriers. Hindrikson et al. (2017) showed that the genetic diversity of wolf populations in Europe could be influenced by other populations up to 850 km away, suggesting differentiation of the wolf populations at a large scale. However, our results show that the northern Iberian wolf population could display differentiation at very much smaller scales in line with observations of Finnish wolves (Aspi, Roininen, Ruokonen, Kojola, & Vilà, 2006; Aspi et al., 2009), which indicate average dispersal distances of about 100 km and significant population differentiation. A study using whole-genome sequences of Scandinavian wolves has also shown large differences in realized inbreeding (as measured by FROH) and heterozygosity among immigrant wolves arriving from the neighbouring Finnish–Russian population (Kardos et al., 2018). The presence of particularly inbred individuals led the authors to suggest that they originated from a small peripheral population. In any case, simulation studies show that the realized inbreeding can vary extensively even among individuals with the same pedigree inbreeding (Hedrick, Kardos, Peterson, & Vucetich, 2017).
Our analyses also show that one of the previously published Iberian wolf genome sequences (wSpain) contained about 14.3% of dog blocks (Table 1). This is close to the 12.5% that would be expected if one of the great-grandparents of this wolf was a dog. However, the haplotype reconstruction shows that dog blocks were fragmented into a very large number of small fragments (Figure 4b) and the ROHs tended to be short (Supporting Information Figure S4). Considering that the number of crossovers per chromosome arm in dogs and wolves is between 1.00 and 1.28 (Muñoz-Fuentes et al., 2015), it seems unlikely that the large number of blocks observed could have been generated by recombination in just three generations. Instead, our results suggest that the presence of small dog chromosome blocks may be compatible with ancient interbreeding events (Thompson, 2013) and imply that genes of dog origin might be pervasive within the genomes of Iberian wolves. Previous studies have suggested that some dog genes may have been introgressed and been positively selected for and spread in certain North American and Eurasian wolf populations (Anderson et al., 2009; Pilot et al., 2018). However, our study shows that the introgression of a very large number of dog chromosomal fragments may not be as rare as initially thought.
Interestingly, the study of a small panel of autosomal microsatellites using structure, one of the most common approaches used to assess introgression and hybridization in natural populations, failed to indicate any admixture for this same sample (Supporting Information Figure S8). This suggests that the power to identify introgression in many other studies may have been limited because of the low number of markers or due to the generalized introgression of one gene pool into the other (see Sanchez-Donoso et al., 2014). Consequently, previous estimates of introgression rates in European wolf populations should be taken with caution (Godinho et al., 2011; Pacheco et al., 2017; Verardi, Lucchini, & Randi, 2006; Vilà, Sundqvist, et al., 2003; Vilà, Walker, et al., 2003). A frequent pattern emerging from most of these studies was the identification of a larger number of F1 hybrids compared to a small proportion of later-generation back-crosses (that would indicate the introgression of dog genes into the wolf population). This was used to argue that F1 hybrids had low success back-crossing into the wolf population and, consequently, they may have a minor effect on the gene pool of wolves. However, our results suggest that this may not be the case and that introgression of dog alleles have occurred repeatedly and may have been underestimated with common analytical approaches.
4.2 Allee effects in the Sierra Morena wolf population
Wolf population size estimates in the Iberian Peninsula are difficult due to the lack of a winter snow cover over most of the range, which makes monitoring very challenging. The estimates of the population size are thus based on the location of family groups and the confirmation of reproduction using direct or indirect approaches (Blanco, Reig, & de la Cuesta, 1992). The difficulties for a comprehensive survey of the wolf population are even larger for the southern population due to the difficult access to most of the presumed range. This has resulted in very few population surveys. Despite this, available reports from the regional government indicate a precipitous decline in the last decades. The wolf sample from Sierra Morena that we studied seems to confirm this bleak view. The number of packs seems to have been under 10 and decreasing for the last 30 years or more. The wolf sample that we studied, road-killed in 2003, shows a level of inbreeding higher than expected for full siblings and lower just to that observed in the inbred Italian wolves (which went through a dramatic bottleneck during the 20th century [Chapron et al., 2014]) and Mexican wolves (captive, derived from a handful of founders; Hedrick & Fredrickson, 2008), and consistent with a small and isolated population. However, as the information about this population is so scarce, there is no direct evidence of inbreeding depression.
Almost a third of the genome of this wolf had dog blocks. This is more than expected if just one of the grandparents was a dog and may be consistent with recurrent hybridization events, where recent hybridization (as shown by the large size of many of the dog blocks in Figure 4a) complemented ancient intercrossing events and introgression of dog genes. This could be an example of Allee effect in an extremely small wolf population. In southern Norway, the presence of a lone female wolf resulted in the only hybridization event reported so far in Scandinavia (Vilà, Sundqvist, et al., 2003; Vilà, Walker, et al., 2003). Similarly, disturbance of the social structure and wolves at the edge of the distribution seem to explain most of the wolf–dog hybridization events in Europe (Andersone, Lucchini, & Ozoliņš, 2002; Godinho et al., 2011; Leonard, Echegaray, Randi, & Vilà, 2014; Verardi et al., 2006). The lack of a stable and large enough wolf population in Sierra Morena may have favoured hybridization with dogs, which are very intensively used in large packs for hunting deer in the area and may frequently escape from human control.
Surprisingly, the high proportion of dog blocks in the genome of the Sierra Morena wolf did not result in an increased heterozygosity (Table 1). In principle, such crosses could be expected to facilitate the genetic rescue of inbred populations with low genetic variation (Hedrick & Fredrickson, 2008; Hedrick & Garcia-Dorado, 2016). However, the genetic diversity in this individual was the lowest among Iberian wolves. The reason for this was that most of the large dog blocks found in one chromosome paired with similar dog blocks in the complementary chromosome (Dog/Dog blocks) as a consequence of the high recent inbreeding in this population resulting in identity by descent. Very large ROHs were detected and almost entire chromosomes were under the homozygosity threshold (Figure 1), as observed in Scandinavian wolves that have suffered very high inbreeding for the last 10 generations (Kardos et al., 2018). The low heterozygosity observed in Wolf/Wolf blocks for this individual, around 1 heterozygous position per 1,000 bp, was practically the same as for Wolf/Dog and Dog/Dog blocks (Figure 5). The low heterozygosity in regions of dog ancestry can be explained by the fact that many of them were found as large Dog/Dog blocks, and purebred dogs tend to have a very low genetic diversity compared to wolves. The complete disappearance of large blocks of wolf chromosomes in Sierra Morena could also be explained by a higher genetic load that could result in selection favouring dog blocks as observed by (Anderson et al., 2009) for melanistic wolves in North America. However, this seems unlikely because the population size for this population was exceedingly small and selection would be inefficient to result in these dramatic changes in the genome structure (Whitlock, 2000).
The results of this study for Sierra Morena wolves are based on the analysis of one single genome. Given the variation in individual ancestry and in realized inbreeding and heterozygosity that could be expected in individuals with the same pedigree (Hedrick et al., 2017), our results may seem to offer a biased view on a population's history. However, this population has just included one or very few packs in the last decades and the studied sample could represent one of the last individuals in the population.
Overall our results show the synergistic effect of diverse Allee effects on this wolf population. As the population declined to a very small size towards the end of the last century, inbreeding increased reaching levels higher than expected for the offspring of matings between full siblings. At the same time, the reduced population size resulted in diminished chances to find mates of the same species, leading to hybridization with dogs. Despite this, the population size did not increase and high inbreeding continued for the hybrid population, trapping the population in an extinction vortex (Gilpin & Soulé, 1986). A wolf census has been carried out across Spain in recent years (MAGRAMA, 2016), but extensive surveys in Sierra Morena failed to find any evidence of wolf breeding. The Sierra Morena wolf population may now be extinct.
5 CONCLUSIONS
Several lessons can be learned from the likely disappearance of the Spanish Sierra Morena wolf population. First, the persistence of a very small population during multiple generations cannot be taken as a sign of viability, as also exemplified by the collapse of the Isle Royale wolf population after decades of inbreeding (Hedrick et al., 2014) and the growing evidence of severe inbreeding effects in small isolated wolf populations (see above). Second, Allee effects are multifaceted and act synergistically magnifying, threats to small populations. Allowing populations to grow until reaching a minimum size may be the best protection against these multiple threats. Third, the introgression of dog alleles into wolf populations may be more pervasive than commonly deduced from studies involving a small number of markers. However, this does not imply that the gene flow is likely to result in a hybrid swarm. Divergence despite genetic exchange is a well-known process (Arnold, 2015) and Iberian wolves are likely to have coexisted with numerous dogs for millennia but remain clearly differentiated and the wolves continue playing an important ecological role. Consequently, the lethal control of putative hybrids suggested in many management plans across Europe [see Boitani (2000) and national and regional Action Plans] may not be needed and, in fact, the perturbation to the wolf populations induced by the lethal control could promote further hybridization (Andersone et al., 2002; Leonard et al., 2014).
ACKNOWLEDGEMENTS
The authors thank J.M Kidd (University of Michigan Medical School) for early access to EEP genome data and Adam Boyko for providing the 48K genotypes. Mario Quevedo and Jorge Echegaray assisted in preparing the distribution map in Supporting Information Figure S1. This project was supported by a project awarded to OR by Fundació Barcelona Zoo and Ajuntament de Barcelona (Spain). We thank the members of the Conservation and Evolutionary Genetics Group at the Doñana Biological Station (EBD-CSIC) for valuable discussions and Belen Lorente-Galdos for feedback on analyses and manuscript.
DATA ACCESSIBILITY
WGS information for wSierraMorena was submitted to the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database under the Accession no. PRJNA482523. Other samples are available in the NCBI SRA database under the Accession Nos. SRP073312 (Botigué et al., 2017), SRA068869 (Wang et al., 2013), SRP044399 (Fan et al., 2016) and PRJNA274504 (Freedman et al., 2014). Genotypes for the 48K data set (Boyko et al., 2010; vonHoldt et al., 2010, 2011) were provided by Adam Boyko.
AUTHOR CONTRIBUTIONS
D.G.S., B.L.G., C.E., T.M.-B., C.L.-F., R.K.W., J.A.L., C.V. and O.R. contributed to the design of this research. D.G.S., I.O., N.S. and O.R. performed the experimental analyses. D.G.S, I.O. and B.L.G. performed the data analysis. D.G.S., J.A.L., C.V. and O.R. wrote the manuscript. All authors read and approved the final manuscript.

