

Low-temperature Spectroscopy of Met100Ala Mutant of Photoactive Yellow Protein†‡
This invited paper is part of the Symposium-in-Print: Photoreceptors and Signal Transduction.
This paper is dedicated to Professor Fumio Tokunaga on the occasion of his retirement from Osaka University.
Abstract
The trans-to-cis photoisomerization of the p-coumaroyl chromophore of photoactive yellow protein (PYP) triggers the photocycle. Met100, which is located in the vicinity of the chromophore, is a key residue for the cis-to-trans back-isomerization of the chromophore, which is a rate-determining reaction of the PYP photocycle. Here we characterized the photocycle of the Met100Ala mutant of PYP (M100A) by low temperature UV–visible spectroscopy. Irradiation of M100A at 80 K yielded a 380 nm species (M100ABL), while the corresponding intermediate of wild type (WT; PYPBL) is formed above 90 K. The amounts of redshifted intermediates produced from M100A (M100AB′ and M100AL) were substantially less than those from WT. While the near-UV intermediate (PYPM) is not formed from WT in glycerol samples at low temperature, M100AM was clearly observed above 190 K. These alterations of the photocycle of M100A were explained by the shift in the equilibrium between the intermediates. The carbonyl oxygen of the thioester linkage of the cis-chromophore in the photocycle intermediates is close to the phenyl ring of Phe96 (<3.5 Å), which would be displaced by the mutation of Met100. These findings imply that the interaction between chromophore and amino acid residues near Met100 is altered during the early stage of the PYP photocycle.
Introduction
Photoactive yellow protein (PYP), first isolated from a purple photosynthetic bacterium Halorhodospira halophila (1), is an excellent target for investigating the light-capturing mechanism of photoreceptor proteins at the atomic level (2,3). PYP is a putative photosensor protein for the negative phototaxis of the bacteria (4). PYP is a water-soluble protein composed of 125 amino acid residues, and its tertiary structure at high resolution is available (5–7). It is a typical PAS domain protein, composed of a six-stranded β-sheet and several short α-helices. To absorb visible light, the trans-p-coumaroyl chromophore binds to Cys69 by a thioester bond (8–10). A photon absorbed by the chromophore induces the trans-to-cis isomerization of the vinyl bond, which initiates a photocycle similar to that of bacterial retinal proteins. The PYP photocycle comprises several intermediates, as demonstrated by previous low-temperature spectroscopy studies (11,12). Blue light or yellow light converts PYPdark (λmax is 449 nm in glycerol samples) into PYPB (489 nm) or PYPH (442 nm), respectively, at 80 K. Both of them are thermally converted to PYPL (456 nm), which is likely to be the same species as I1 (=pR) formed on the microsecond time scale at room temperature (13,14). At room temperature, PYPL is converted to a near-UV intermediate (PYPM, I2 or pB) (356 nm) as a consequence of a protein conformational change (15–17), but in glycerol samples at low temperature, PYPL is directly converted to the dark (ground) state without the formation of PYPM.
Using site-directed mutants, the roles of various amino acid residues of PYP in the structure and photoreaction have been studied so far. One of the key amino acid residues governing the PYP photocycle is Met100 (18–20), which is located in the loop region connecting the fourth and fifth β-strands (Met100 loop). This region is curled toward the chromophore, and the distance between the chromophore and Met100 side chain is less than 4.5 Å in the dark state (PDB ID 1nwz) (Fig. 1). Mutation of Met100 substantially slows the photocycle (18–20). M100A shows much slower decay of the I2 state (M100AM), whereas the decay of the I1 state (M100AL) is faster than that of the wild type (WT) (18). As the back-isomerization of the cis-chromophore in M100AM by light rapidly generates M100ADark, Met100 is thought to catalyze the cis-to-trans isomerization of the chromophore during the photocycle (18).

Spatial relationship between photoactive yellow protein chromophore and nearby amino acid residues (PDB ID 1nwz) (6).
While M100AM has been extensively characterized, the primary photoreaction of M100A has not been studied in detail. Prof. Fumio Tokunaga and his coworkers have demonstrated that low temperature spectroscopy is a powerful tool to analyze the photoreaction and subsequent thermal reactions of photoreceptor proteins. Using liquid nitrogen as a coolant, one can observe the ultrafast reactions that take place on the picosecond time scale at room temperature using a conventional UV–visible recording spectrophotometer. Heating the sample mimics the elapse of time. Thus, the whole reaction can be easily and consistently covered using a single experimental setup. Here, the photocycle of M100A was studied by low-temperature spectroscopy to clarify the role of Met100 in the early stage of the PYP photocycle.
Materials and methods
Apoproteins of WT PYP and M100A were prepared according to the standard protocol (21). They were reconstituted into holoproteins and purified by DEAE-Sepharose column chromatography as reported previously (10,21). WT and M100A were suspended in 10 mm TAPS buffer at pH 8.0 containing 200 mm NaCl. Two volumes of glycerol were added to the sample for low-temperature spectroscopy (66% glycerol sample).
The sample was put into a sample cell composed of a transparent quartz window, an opal glass window (Sigma Koki, Tokyo, Japan) and a silicon rubber spacer (2 mm in thickness, 6 mm in inner diameter) (22). It was set in an optical cryostat (Oxford Optistat DN) installed in a recording spectrophotometer (Shimadzu UV2400PC). The reference beam of the spectrophotometer was similarly scattered with another opal glass plate. The slit of the measurement beam was set at 2 nm. The sample was irradiated with blue (436 nm) or yellow (>450 nm) light passed through an optical interference filter (Edmund 43161) or a glass cutoff filter (Asahi Techno Glass Y47). A 1-kW slide projector was used as an irradiation light source.
Wild type and M100A were irradiated at 80 K and the subsequent thermal reaction of the photoproduct was induced by warming. The temperature of the sample, which was monitored by the thermocouple attached to the sample cell holder, was increased at 1 K min−1 using an ITC502 temperature controller (Oxford). The absorption spectra were recorded every 10 K.
The absorption spectra of PYP and intermediates are sharpened by cooling (11). Therefore, increasing the temperature induces thermal broadening of the absorption spectra as well as thermal decay of the intermediates. In the previous low temperature spectroscopy, the sample, warmed to induce the thermal reaction, was re-cooled to the base temperature (83 or 193 K) for measurement to avoid the effect of temperature on the absorption spectrum (11). However, repetition of warming and cooling readily shifts the baseline. In the present experiment, the absorption spectrum of the dark state was measured every 10 K, and the difference spectra before and after irradiation were calculated using the dark spectrum and light spectrum measured at the same temperature to minimize the effect of temperature on the absorption spectra.
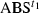 ) is composed of the spectra of intermediate(s) present at t1 (
) is composed of the spectra of intermediate(s) present at t1 (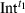 ) and dark state at t1 (
) and dark state at t1 (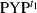 ) as follows.
) as follows.
 (1)
(1)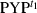 was the major component. In the present analysis,
was the major component. In the present analysis, 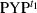 was subtracted from
was subtracted from 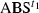 , giving the difference spectrum (Diff) as follows:
, giving the difference spectrum (Diff) as follows:
 (2)
(2)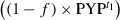 is canceled, and the shape of the difference spectrum is independent of f.
is canceled, and the shape of the difference spectrum is independent of f.Results
Photoreaction at 80 K
The absorption maximum of M100A in the visible region is located at 446 nm, and it is shifted to 448 nm in 66% glycerol buffer. A similar small redshift by glycerol at high concentration is also observed for WT (11). The absorption spectrum of M100A at 80 K was sharpened and showed a spectral shoulder at 420 nm, which was similar to the sharpening and shoulder observed for WT (Fig. 2). WT and M100A in 66% glycerol buffer were irradiated with blue light (436 nm) or yellow light (>450 nm) at 80 K and the absorption spectra were recorded (Fig. 2).

Photoreaction of wild type (WT) and M100A at 80 K. WT (top) or M100A (bottom) suspended in 66% glycerol buffer was cooled to 80 K and the spectrum was recorded (curve 1 for WT and curve 4 for M100A). It was irradiated with blue light (436 nm) for 240 s (curves 2 and 5) or yellow light (>450 nm) for 240 s (curves 3 and 6). Inset—The spectral changes induced by blue and yellow light were calculated by subtracting dark-state spectra from the respective spectra.
Irradiation of WT with blue light yielded a significant amount of the redshifted intermediate called PYPB, which corresponds to I0 at room temperature (23,24) (Fig. 2, curve 2). After irradiation for 240 s, no further spectral shift was observed upon prolonged irradiation because the photo-steady-state mixture was produced. On the other hand, yellow light caused a 7 nm blueshift in the absorption maximum, indicating the formation of a blueshifted intermediate called PYPH (curve 3). As the extinction coefficient of PYPH is smaller than that of PYPdark throughout the visible region, no absorbance increase was observed in the difference spectrum between them (curve 3′ in inset). Due to the shift in the peak and spectral shoulder, the difference spectra between PYPH and PYPdark showed two sharp negative bands (curve 3′ in inset). The presence of a pair of sharp negative bands in the difference spectra is a marker of the formation of PYPH. Thus, the photo-steady-state mixture produced by blue light contained PYPH as well as PYPB (curve 2′ in inset).
Irradiation of M100A with blue light produced a blueshifted intermediate having a maximum at 380 nm (curve 5 in Fig. 2). The spectroscopic properties of this photoproduct were similar to those of PYPBL (M100ABL). Concomitantly, a small amount of redshifted intermediate was formed (curves 5 and 5′). However, the absorbance increase at 490 nm indicating the formation of this product (M100AB′) was much smaller than that of PYPB. In addition, the absorption band of M100 AB′ was located in a wavelength region 20–30 nm shorter than that for PYPB. The broad negative band without a pair of sharp negative bands (curve 5′) shows that the amount of M100AH in the photo-steady-state mixture produced by the blue light was smaller than that of PYPH.
Irradiation of M100A with yellow light induced a blueshift in the absorption spectrum (curve 6). The absorbance increase at 390 nm and a pair of negative bands in the difference spectrum (curve 6′) showed the formation of M100ABL and M100AH, respectively.
Thermal reactions of photoproducts formed at 80 K
The photo-steady-state mixture produced by blue light or yellow light at 80 K was warmed at a steady rate of 1 K min−1, and the absorption spectra were recorded at every 10 K. Because the mixture was not equilibrated, the spectra of the transient mixture were recorded. The dark-state spectra measured at the same temperature were then subtracted to obtain the difference spectra between the dark state and the photocycle intermediate(s) (Fig. 3). Prior to examining M100A, the photocycle of WT was surveyed to validate the present experimental setup and analysis (broken lines in Fig. 3). Irradiation of WT with yellow light at 80 K yielded PYPH, as shown by the pair of sharp negative bands and absence of a positive band (top of left-hand column). As a result of warming, the negative band turned broad, indicating the formation of PYPHL (123–163 K) (11) and the redshifted intermediate (PYPL) was formed above 183 K. At 213 K, PYPL reverted to the dark state without the formation of the near-UV intermediate (PYPM).

The thermal reaction of photoproducts formed from M100A. The photo-steady-state mixture produced by >450 nm light (left-hand column) or 436 nm light (right-hand column) was warmed at the rate of 1 K min−1 and the absorption spectra were recorded every 10 K. The dark-state spectra measured at the same temperature were subtracted to show the difference between thermal product and dark state. The temperatures of the measurements are indicated in the figure. For comparison, the difference spectra for wild type measured under the same condition are shown by broken lines.
The mixture of PYPB and PYPH produced by blue light was similarly warmed (right-hand column). Warming the photo-steady-state mixture caused the decay of PYPB to PYPBL, which was completed at 133 K. At this temperature, the negative band was broadened due to the conversion from PYPH to PYPHL, as shown in the left-hand column. PYPBL was converted to PYPL above 173 K, and then PYPL directly reverted to the dark state. These spectral changes were consistent with the previous low temperature spectroscopy in which the effect of temperature on the absorption spectrum was avoided by measuring at fixed temperature after warming (11).
The thermal reactions of the photoproducts produced from M100A were studied in the same manner (solid lines in Fig. 3). The photo-steady-state mixture produced by yellow light, which contained M100ABL and M100AH, was warmed (left-hand column). The sharp negative bands were first broadened (123–163 K), and then M100AL was formed (183 K) and decayed above 193 K. Concomitant with the decay of M100AL, the positive 380 nm band was shifted to 360 nm, showing that M100ABL was converted to M100AMvia M100AL (193–203 K).
M100AB′ formed at 80 K showed a 20–30 nm blueshifted spectrum relative to that of PYPB (right-hand column). Along with M100AB′, M100ABL was formed at 80 K by blue light. Upon warming, M100AB′ decayed at 113 K, and M100AL was formed at 183 K. While the spectral properties of M100AB′ and M100AL were very similar to each other, the decay and formation of the 480 nm species clearly showed that they are distinct species (see Fig. 4). At 193 K, M100AL was converted to M100AM. It should be noted that PYPBL was completely converted to PYPL (difference spectra of WT at 193–203 K), whereas M100ABL was present over a wide temperature range (83–203 K). The 380 nm band of M100ABL was shifted to 360 nm, indicating the formation of M100AM, at 223 K. The difference spectra at 233 K clearly showed the formation of M100AM, while PYPM was not observed in this condition.

Heat-induced absorbance changes at typical wavelengths. The difference absorbance at 485, 388 and 350 nm for wild type (WT) and 485, 385 and 325 nm for M100A in light/dark spectra (Fig. 3) was plotted against the temperature.
To illustrate the thermal behavior of intermediates, the heat-induced absorbance changes at typical wavelengths are presented (Fig. 4). For WT, the absorbance at 485 nm indicates the presence of PYPB and PYPL, whereas those at 388 and 350 nm indicate that of PYPBL. The absorbance decrease at 485 nm is complementary to the increase at 388 or 350 nm and vice versa, indicating a simple sequential reaction. However, M100ABL was present over a wide temperature range despite the decay and formation of M100AB′ and M100AL, showing the equilibrium between M100ABL, M100AB′ and M100AL. The photocycle of M100A is illustrated in Fig. 5 in comparison with that of WT.

Photocycles of wild type and M100A at low temperature.
Discussion
In the present study, the photocycle of M100A was studied by low-temperature UV–visible spectroscopy. M100A was irradiated at 80 K and the subsequent thermal reactions were induced by warming the sample. Warming caused thermal broadening of the absorption spectra as well as decay of the thermolabile species. The percentages of the photocycle intermediate(s) contained in the photo-steady-state mixture produced at 80 K were estimated to be 30% for blue light and 10% for yellow light. Therefore, the effect of temperature on the absorption spectrum of the major component (dark state) is not negligible. To minimize the effect of temperature on the dark-state spectrum, the difference spectra were calculated by subtracting the dark-state spectra measured at the same temperature (Eq. 2). In this method, the effect of temperature on the absorption spectra was small enough to follow the decay-associated spectral changes (Fig. 3).
Above 213 K, PYPL in 66% glycerol buffer directly reverted to PYPdark, indicating that the cis-to-trans back-isomerization takes place more readily than the proton transfer from the chromophore to Glu46 (25,26). In contrast, M100AL decayed at lower temperatures than PYPL and was converted to M100AM, suggesting that the equilibrium between M100AL and M100AM (27,28) is shifted toward the latter. It is well known that the decay of M100AM at room temperature is extremely slow, while I1 of M100A (M100AL) decays very fast (18). Therefore, the lower decay temperature of M100AL compared to PYPL is consistent with the findings at room temperature.
The difference spectra obtained by low-temperature spectroscopy of M100A before and after irradiation were quite different in shape from those of WT. However, the photocycles of M100A and WT were explained by qualitatively similar intermediates by taking the thermal equilibrium into consideration (Fig. 5). The most marked difference was the presence of M100ABL over a wide temperature range. For WT, PYPBL is not formed at 80 K—it is thermally formed from PYPB above 93 K. The apparent decay temperature of M100AB′ was 20 K higher than that of WT. If the back reaction from M100ABL to M100AB′ does not take place, M100AB′ should be more stable than PYPB. However, M100ABL formed at a lower temperature than PYPBL, indicating the presence of equilibrium between M100AB′ and M100ABL. In addition, the decay of M100ABL and M100AL took place simultaneously. Therefore, M100AB′, M100ABL and M100AL are in thermal equilibrium, which favors M100ABL (Fig. 5).
We previously proposed that the progress of the photocycle from PYPB or PYPH to PYPL is a consequence of the relaxation of the twisted double bond of the chromophore (29). The photo-isomerization of the chromophore takes place by flipping the carbonyl oxygen (O1) of the thioester bond (22,25,29–31) (Fig. 6). Although the thermal equilibria between the photocycle intermediates are markedly shifted by mutation of Met100, it is unlikely that Met100 directly interacts with the chromophore, because the distance between C3 of the chromophore and S of Met100 is 4.10 or 4.51 Å in PYPB (PDB ID 1uwp) (30) and 4.53 Å in PYPBL (3pyp) (31). On the other hand, trans-to-cis isomerization moves O1 toward the phenyl ring of Phe96, which may cause steric hindrance. In addition, electrostatic repulsion would be present between O1 and the π-electron system of the phenyl ring. Thus, it is likely that PYPB, PYPBL, PYPH, PYPHL and PYPLdiffer as regards to the interactions between O1 and Phe96. S of Met100 and N of Arg52 are located at a distance of 3.88 Å (PDB ID 2D01), suggesting weak interaction via the NH-S hydrogen bond (32). Mutation of Met100 eliminates this interaction, resulting in a structural change in the Met100 loop. As a consequence, M100ABL would be stabilized by the displacement of the side chain of Phe96. At low temperatures, the reverse reaction from PYPL to PYPdark is dominant over the conversion to PYPM, and would also be driven by the interaction between O1 and Phe96.

The crystal structures of PYPdark and PYPB. The cryotrapped structures of PYPdark (yellowish) and PYPB (reddish) are superimposed (PDB ID 1uwp) (30). Note that Met100 (selenomethionine) is in double conformation in the dark.
Mutation of Phe96 substantially slows the decay of PYPM. The decay time constant of F96A is 1000 s (33), which is comparable to that of M100AM. While this suggests the occurrence of the interaction between O1 and Phe96 in PYPM, it is not likely that the spatial relationship between the chromophore, Phe96 and Met100 demonstrated by the crystallography of PYPB is maintained in PYPM, because the Met100 loop undergoes a large conformational change (34–36) that has not been observed by crystallography (37). Thus, the mechanism by which Met100 catalyzes the cis-to-trans isomerization remains unclear because of the lack of the high-resolution structure of PYPM in the physiologic condition. The lifetimes of M100AM, M100KM and M100LM are 1000 times longer than that of PYPM, but M100EM decays 10 times faster than M100AM (20), suggesting that the proton acceptor for the hydrogen bond at position 100 facilitates the decay of PYPM. Thus, the NH-S hydrogen bond between Met100 and Arg52 partially contributes to the decay of PYPM, as indicated by the fact that the lifetime of R52QM, in which the distance between S of Met100 and N of Gln52 is 8.57 Å in the dark state (PDB ID 2D02) (38), is five times longer than that of PYPM.
Acknowledgments
Acknowledgements— We thank Dr. Elizabeth Nakajima for critical reading of the manuscript. This research was financially supported in part by the Global Center of Excellence Program “Formation of a Strategic Base for Biodiversity and Evolutionary Research: from Genome to Ecosystem” of the Ministry of Education, Culture, Sports and Technology (MEXT), Japan.

