

Interactions Between Chromophore and Protein in Phytochrome Identified by Novel Oxa-, Thia- and Carba-Chromophores
Abstract
Six new bilin chromophores of the plant photoreceptor phytochrome have been synthesized, carrying at the photoisomerizing ring D an oxygen or a sulfur atom or a methylene group instead of the pyrrole nitrogen atom. These furanone-, thiophenone- or cyclopentenone-containing compounds bound covalently to the recombinant apophytochrome phyA of Avena sativa. The novel chromoproteins showed hypsochromically shifted absorption spectra with respect to native phytochrome and a strongly diminished photochemical activity, but a three- to four-fold higher fluorescence quantum yield. These results demonstrate that, on the one hand, also ring D-modified chromophores can be forced into a partially extended structure, required for incorporation into the apoprotein binding pocket and covalent binding. On the other hand, the modifications introduced into ring D of the chromophores strongly impede the formation of stable far red-absorbing forms of plant photoreceptor phytochrome (Pfr-form) of the chromoproteins, highlighting especially the role of the pyrrole nitrogen atom and hydrogen bonding for the precise interactions between that part of the chromophore and the protein for the Pfr-formation.
Introduction
Phytochromes are master regulators for plant photomorphogenesis (1). They gain their capability as signal transducing proteins from a tight interaction between the protein moiety and their covalently bound chromophore, a bilin compound that undergoes a photoisomerization of one of its double bonds connecting the four pyrrole rings [C(15)–C(16), Fig. 1]. In most plant species investigated so far, phytochromobilin (PΦB, #1), is found as chromophore, except in two algal species, where phycocyanobilin (PCB, #2), has been identified (2,3). The finding of phytochrome-like proteins also in a large number of prokaryotic species (4), and a recently presented three-dimensional structure of a chromophore-binding domain from the phytochrome of Deinococcus radiodurans have unveiled quite a number of interactions between the chromophore and the surrounding protein. In the prokaryotic phytochromes, two classes of chromoproteins can be discerned: one using PCB (2) as chromophore, which binds covalently to a conserved cysteine in the cGMP-specific and -regulated cyclic nucleotide phosphodiesterase, adenylyl cyclase and E. coli transcription factor FhlA protein domain (GAF) as in the plant phytochromes, the other class employing biliverdin (BV) IXα, which is attached to a cysteine at the front end in the Per-Arnt-Sim (PAS, GAF, abundantly found protein folding motifs) domain. Although the phytochrome from D. radiodurans binds BV, the crystal structure revealed quite a number of hydrogen bonds and electrostatic interactions between the chromophore and many amino acids that are highly conserved in most other phytochromes, allowing to propose a generalized mechanism of chromophore fixation and directed photoisomerization. The stabilization of the far red-absorbing forms of plant photoreceptor phytochrome (Pfr-state), however, remains uncertain: the protein pocket around the photoisomerizing ring D of the chromophore is of fairly hydrophobic structure, and also ring D does not exhibit significantly functionalized substituents that might exert specific interactions to stabilize the Pfr-form after its formation. We thus proposed that it might be the pyrrole nitrogen atom itself and the attached proton that play an important role for the Pfr-stability. Based on a recent synthetic approach (5), we here present the six novel phytochrome chromophores 4–9 which instead of the photoisomerizing pyrrole ring D carry a furanone, a thiophenone, or a cyclopentenone ring, in each case substituted with either 18-ethyl-17-methyl or 17,18-dimethyl substituents (Fig. 1). The role of the modified ring D structure in chromoprotein generation and in their spectral properties is discussed.

The upper Kekulé formula shows PΦB (1) and PCB (2) in the ZZZ,asa state. Both chromophores vary from each other exclusively in their substitution pattern at position C(18) (vinyl vs ethyl) and are covalently bound to a conserved cysteine (position 321 of oat phytochrome) via a thioether linkage. The photoisomerization of the C(15)–C(16) double bond is indicated by an arrow. The lower Kekulé formula shows the structure of the PCB-analogous 4–9 which differ from the native chromophores 1 and 2 in their substitution pattern at ring D. For reasons of clarity they are shown with the same stereochemistry as the protein-bound chromophores 1 and 2.
Materials and methods
Biochemical section.
General methods: Generation of recombinant apophytochrome (N-terminal 65 kDa domain of oat phyA) followed essentially formerly reported literature protocols (6). Sample treatment was at ambient temperature and spectroscopic measurements were performed under temperature-controlled conditions at 20°C. Assembly of chromoproteins was accomplished by addition of 4–9 to an aqueous, buffered solution (50 mm Tris, 300 mm NaCl and 5% glycerol, pH 7.5) of the recombinant apophytochrome. Absorption spectra were recorded after the addition and purification of the respective chromoprotein at short time intervals for a period of 72 h. Measurements were done in the same buffer as used for the assembly reaction. After assembly, samples were handled under safe light (450–500 nm). Irradiation of the novel phytochromes was performed with light from a 100 W slide-projector bulb, equipped with appropriate interference filters [IF; irradiation of the Pr-, Pfr-form: 4- and 5-containing phytochromes: 616 nm (IF), 655 nm (IF); 6- and 7-containing phytochromes: 578 nm (IF), 715 nm (cut-off filter; CF); and 8- and 9-containing phytochromes: 578 nm (IF), 654 nm (CF); all IFs had a half-bandwidth of ±7 nm]. Samples were irradiated at a distance of 16 cm. A sample of PCB-assembled phytochrome was always used as control. Chromophore competition experiments were performed such that 2 μL of a concentrated solution of PCB (2) in DMSO was added to the fully assembled analog chromoprotein. UV/Vis: Shimadzu UV-2401 PC spectrometer. Fluorescence: Varian Cary Eclipse spectrometer. Data-acquisition and -treatment was performed with the OriginPro 7.5 software.
Chemical section.
General methods:
Column chromatography: Purifications were performed on Merck silica gel 60 (230–400 mesh) under slightly increased pressure.
HPLC: Purifications were carried out on the following columns: Kromasil C18 ODS-5-100 (RP-C18, 5 μm, 250 × 21 mm, flow rate 0.8 mL min−1) (column A), Phenomenex Luna 5μ C18 (RP-C18, 5 μm, 125 × 4.6 mm, flow rate 1.0 mL min−1) (column B), Phenomenex Gemini 5μ C18 (RP-C18, 5 μm, 150 × 4.6 mm, flow rate 1.2 mL min−1) (column C) or GLS Sciences, Inc. Inertsil C8-3 (RP-C8, 5 μm, 125 × 4.6 mm, flow rate 0.8 mL min−1) (column D).
NMR: Spectra were recorded on Bruker ARX 250, DRX 400 or DRX 500 spectrometers in the solvents indicated; chemical shifts (δ) are given in ppm relative to TMS, coupling constants (J) in Hz. The solvent signals were used as references and the chemical shifts were converted to the TMS scale (CDCl3: δC = 77.0 ppm; residual CHCl3 in CDCl3: δH = 7.24 ppm; pyridine-d5: δC = 123.5 ppm; residual pyridine in pyridine-d5: δH = 7.19 ppm). IR: Perkin-Elmer System 2000 spectrometer; wavenumbers 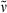 in cm−1.
in cm−1.
UV/Vis: Shimadzu UV-2401 PC spectrometers; the solvents applied were of appropriate quality (Uvasol). MS (EI): Finnigan MAT 8200 or MAT 8400 (70 eV); ESI-MS: Bruker Esquire 3000 or HP Quadrupol MS Engine; accurate mass determinations (HRMS): Finnigan MAT 95; MALDI-TOF-MS: Applied Biosystems Voyager DE-Pro spectrometer connected with a LeCroy high speed digitizer (20 kV acceleration voltage, 130 ns delay time). Melting points: Reichert melting point microscope (uncorrected).
Although the nomination of the aldehydes 16–21 correlates with the IUPAC rules, for prudential reasons, the assignment of the hydrogen and carbon atoms in the case of NMR-analysis follows the Kekulé formula displayed in Fig. 2.

Synthesis of the aldehydes 16–21 which represent the “right halves” of novel PCB-like chromophores 4–9.
More detailed information concerning the reaction conditions are available directly from the authors.
Methyl 3-{5-[(4-ethyl-3-methyl-5-oxofuran-2(5H)-ylidene)methyl]-2-formyl-4-methyl-1H-pyrrol-3-yl}propanoate (16). A solution of the CD building block 10 (118 mg, 0.29 mmol) in TFA (8.4 mL, 108 mmol) was stirred for 3 min at ambient temperature in the dark and under argon. After addition of orthoformic trimethyl ester (8.4 mL, 75.2 mmol), the solution was stirred for additional 30 min and subsequently absorbed in methylene chloride (450 mL). This mixture was washed with H2O (2 × 200 mL) and with a saturated solution of NaHCO3 (2 × 200 mL). Afterwards, the organic layer was dried over Na2SO4 and the solvent was evaporated in vacuo. HPLC (column A) of the remaining green crude product with methanol and H2O (5:1) as eluent yielded the expected reaction product 16 (52 mg, 0.16 mmol, 54%) as a yellow-colored solid. m.p. 129°C; 1H NMR (400 MHz, CDCl3) δ = 1.13 (t, 3J = 7.59 Hz, 3H, 92-CH3), 2.11 (s, 3H, 41-CH3), 2.13 (s, 3H, 81-CH3), 2.39 (q, 3J = 7.47 Hz, 3H, 91-CH2), 2.56 (t, 3J = 7.63 Hz, 2H, 32-CH2), 3.03 (t, 3J = 7.63 Hz, 2H, 31-CH2), 3.64 (s, 3H, CO2CH3), 5.89 (s, 1H, 6-CH), 9.69 (s, 1H, CHO), 9.75 (br s, 1H, NH); 13C NMR (101 MHz, 1H/13C-COSY, CDCl3) δ = 8.75 (C-81), 9.67 (C-41), 12.73 (C-92), 17.36 (C-91), 19.11 (C-31), 35.25 (C-32), 51.75 (CO2CH3), 95.07 (C-6), 123.35 (C-3), 129.72, 130.53, 130.68, 132.02 (C-2, -4, -5, -9), 146.54 (C-8), 148.46 (C-7), 169.07 (C-10), 172.78 (CO2CH3), 177.47 (CHO); IR (KBr): 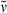 = 3439, 3070, 2965, 2928, 2852, 1759, 1734, 1637, 1444, 1384, 1213, 1164, 1100, 1008, 941, 574 and 458 cm−1; UV/Vis [methanol + H2O (5:1)]: λmax = 266, 394 and 409 nm (ε not determined); MS (EI, 135°C): m/z (%): 331 (100) [M+], 330 (2), 303 (19), 302 (33), 300 (12), 299 (2), 272 (34), 271 (11), 270 (7), 260 (7), 243 (5), 230 (13), 228 (6), 217 (2), 203 (3), 186 (3), 176 (3), 162 (3), 158 (2), 146 (2), 136 (2), 134 (3), 119 (2), 118 (4), 117 (2), 107 (2), 106 (3), 104 (3), 91 (4), 79 (2), 77 (4), 67 (2), 65 (2), 53 (2), 41 (2); HRMS (ESI pos): m/z: calcd for C18H22NO5 [M + H]+: 332.149799; found: 332.14999.
= 3439, 3070, 2965, 2928, 2852, 1759, 1734, 1637, 1444, 1384, 1213, 1164, 1100, 1008, 941, 574 and 458 cm−1; UV/Vis [methanol + H2O (5:1)]: λmax = 266, 394 and 409 nm (ε not determined); MS (EI, 135°C): m/z (%): 331 (100) [M+], 330 (2), 303 (19), 302 (33), 300 (12), 299 (2), 272 (34), 271 (11), 270 (7), 260 (7), 243 (5), 230 (13), 228 (6), 217 (2), 203 (3), 186 (3), 176 (3), 162 (3), 158 (2), 146 (2), 136 (2), 134 (3), 119 (2), 118 (4), 117 (2), 107 (2), 106 (3), 104 (3), 91 (4), 79 (2), 77 (4), 67 (2), 65 (2), 53 (2), 41 (2); HRMS (ESI pos): m/z: calcd for C18H22NO5 [M + H]+: 332.149799; found: 332.14999.
Additionally, the dimerization product 29 (16 mg, 0.022 mmol, 18%) was isolated in the form of a dark blue solid. m.p 178°C; 1H NMR (500 MHz, CDCl3) δ = 1.11 (t, 3J = 7.57 Hz, 6H, 22-, 182-CH3), 2.14 (s, 6H, 71-, 131-CH3), 2.18 (s, 6H, 31-, 171-CH3), 2.35 (q, 3J = 7.50 Hz, 4H, 21-, 181-CH2), 2.53 (t, 3J = 7.48 Hz, 4H, 82-, 122-CH2), 2.92 (t, 3J = 6.46 Hz, 4H, 81-, 121-CH2), 3.64 (s, 6H, CO2CH3), 6.21 (br s, 2H, 5-, 15-CH), 6.83 (br s, 1H, 10-CH); 13C NMR (126 MHz, BB, 1H/13C-COSY, CDCl3) δ = 9.53 (C-71, -131), 10.12 (C-31, -171), 12.41 (C-22, -182), 17.10 (C-21, -181), 19.63 (C-81, -121), 34.90 (C-82, -122), 51.45 (CO2CH3), 98.71 (C-5, -15), 116.14 (C-10), 128.04 (C-7, -13), 137.97 (C-9, -11), 140.76 (C-6), 146.98 (C-3, -17), 148.24 (C-4,- 16), 150.21 (C-14), 169.43 (C-1, -19), 172.83 (CO2CH3); IR (KBr): 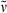 = 3448, 2930, 2874, 1757, 1743, 1640, 1594, 1458, 1437, 1385, 1357, 1260, 1236, 1170, 1097, 1057, 1014, 950, 906, 883, 801, 775, 736, 705, 673 and 613 cm−1; UV/Vis (diethyl ether): λmax (ɛ) = 307 (12 500), 358 (22 900), 596 nm (12 400); MS (EI, 260°C): m/z (%): 616 (100) [M+], 557 (29), 529 (47), 497 (4), 469 (33), 392 (5), 316 (10), 242 (8) and 180 (3); HRMS (ESI pos, methanol + methylene chloride): m/z: calcd for C35H41N2O8 [M + H]+: 617.285743; found: 617.286033.
= 3448, 2930, 2874, 1757, 1743, 1640, 1594, 1458, 1437, 1385, 1357, 1260, 1236, 1170, 1097, 1057, 1014, 950, 906, 883, 801, 775, 736, 705, 673 and 613 cm−1; UV/Vis (diethyl ether): λmax (ɛ) = 307 (12 500), 358 (22 900), 596 nm (12 400); MS (EI, 260°C): m/z (%): 616 (100) [M+], 557 (29), 529 (47), 497 (4), 469 (33), 392 (5), 316 (10), 242 (8) and 180 (3); HRMS (ESI pos, methanol + methylene chloride): m/z: calcd for C35H41N2O8 [M + H]+: 617.285743; found: 617.286033.
Methyl 3-{5-[(3,4-dimethyl-5-oxofuran-2(5H)-ylidene)methyl]-2-formyl-4-methyl-1H-pyrrol-3-yl}propanoate (17): See synthesis of 16. Yellow colored solid (48 mg, 0.15 mmol, 52%). m.p. 152°C; 1H NMR (400 MHz, CDCl3) δ = 1.95 (s, 3H, 91-CH3), 2.11 (s, 3H, 41-CH3), 2.12 (s, 3H, 81-CH3), 2.56 (t, 3J = 7.62 Hz, 2H, 32-CH2), 3.03 (t, 3J = 7.62 Hz, 2H, 31-CH2), 3.64 (s, 3H, CO2CH3), 5.89 (s, 1H, 6-CH), 9.69 (s, 1H, CHO), 9.72 (br s, 1H, NH); 13C NMR (101 MHz, CDCl3) δ = 8.75 (C-81), 8.97 (C-91), 9.89 (C-41), 19.12 (C-31), 35.26 (C-32), 51.71 (CO2CH3), 94.97 (C-6), 123.34 (C-3), 124.27, 126.79, 130.56, 132.41 (C-2, -4, -5, -9), 147.06 (C-8), 148.50 (C-7), 169.17 (C-10), 172.78 (CO2CH3), 177.32 (CHO); IR (KBr): 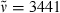 , 3064, 2921, 2854, 1767, 1730, 1633, 1540, 1439, 1396, 1219, 1164, 1094, 1023, 985, 907, 747, 594 cm−1; UV/Vis [methanol + H2O (5:1)]: λmax = 266, 394, 408 nm (ε not determined); MS (EI, 150°C): m/z (%): 317 (100) [M+], 316 (3), 289 (21), 288 (33), 286 (12), 285 (2), 258 (40), 257 (14), 256 (10), 246 (10), 244 (21), 230 (18), 229 (9), 228 (5), 216 (21), 215 (5), 214 (6), 203 (3), 189 (6), 172 (2), 162 (4), 144 (4), 120 (3), 79 (3), 77 (6), 53 (4); HRMS (ESI pos, methanol): m/z: calcd for C17H20NO5 [M + H]+: 318.134149; found: 318.13427.
, 3064, 2921, 2854, 1767, 1730, 1633, 1540, 1439, 1396, 1219, 1164, 1094, 1023, 985, 907, 747, 594 cm−1; UV/Vis [methanol + H2O (5:1)]: λmax = 266, 394, 408 nm (ε not determined); MS (EI, 150°C): m/z (%): 317 (100) [M+], 316 (3), 289 (21), 288 (33), 286 (12), 285 (2), 258 (40), 257 (14), 256 (10), 246 (10), 244 (21), 230 (18), 229 (9), 228 (5), 216 (21), 215 (5), 214 (6), 203 (3), 189 (6), 172 (2), 162 (4), 144 (4), 120 (3), 79 (3), 77 (6), 53 (4); HRMS (ESI pos, methanol): m/z: calcd for C17H20NO5 [M + H]+: 318.134149; found: 318.13427.
Additionally, the dimerization product 30 (13 mg, 0.022 mmol, 16%) was isolated in the form of a dark blue solid. m.p 211°C; 1H NMR (400 MHz, CDCl3) δ = 1.88 (s, 6H, 21-, 181-CH3), 2.11 (s, 6H, 71-, 131-CH3), 2.17 (s, 6H, 31-, 171-CH3), 2.52 (t, 3J = 7.40 Hz, 4H, 82-, 122-CH2), 2.89 (t, 3J = 7.40 Hz, 4H, 81-, 121-CH2), 3.64 (s, 6H, CO2CH3), 6.15 (s, 2H, 5-, 15-CH), 6.75 (s, 1H, 10-CH); 13C NMR (101 MHz, CDCl3) 8.89 (C-21, -181), 9.99 (C-71, 131), 19.87 (C-31, -171), 26.40 (C-81), 29.69 (C-121), 32.20 (C-82), 34.33 (C-122), 51.68 (CO2CH3), 97.20 (C-15), 99.27 (C-5), 115.94 (C-10), 124.00 (C-7), 125.04 (C-2, -18), 128.15 (C-13), 129.65 (C-6, -14), 135.21 (C-9, -11), 141.67, 150.11 (C-8, -12), 147.62 (C-3, -17), 148.57 (C-4, -16), 169.44 (C-1, -19), 173.16 (CO2CH3); IR (KBr):  = 3441, 3065, 2920, 2850, 1744, 1645, 1596, 1459, 1437, 1396, 1350, 1263, 1235, 1171, 1093, 1014, 964, 909, 875, 801, 750, 706, 678 cm−1; UV/Vis (diethyl ether): λmax (ɛ) = 304 (10 300), 358 (20 900), 595 nm (12 400); MS (EI, 235°C): m/z (%): 588 (100) [M+], 587 (5), 573 (5), 557 (16), 556 (19), 529 (36), 514 (2), 501 (46), 482 (3), 469 (5), 465 (2), 441 (44), 428 (33), 405 (2), 383 (2), 359 (3), 357 (2), 332 (3), 331 (4), 319 (5), 294 (4), 289 (2), 287 (2), 274 (3), 262 (2), 257 (3), 241 (4), 240 (3), 228 (4), 227 (5), 221 (3), 220 (2), 57 (2), 43 (2); HRMS (ESI pos, methanol): m/z: calcd for C33H37N2O8 [M + H]+: 589.254992; found: 589.25477.
= 3441, 3065, 2920, 2850, 1744, 1645, 1596, 1459, 1437, 1396, 1350, 1263, 1235, 1171, 1093, 1014, 964, 909, 875, 801, 750, 706, 678 cm−1; UV/Vis (diethyl ether): λmax (ɛ) = 304 (10 300), 358 (20 900), 595 nm (12 400); MS (EI, 235°C): m/z (%): 588 (100) [M+], 587 (5), 573 (5), 557 (16), 556 (19), 529 (36), 514 (2), 501 (46), 482 (3), 469 (5), 465 (2), 441 (44), 428 (33), 405 (2), 383 (2), 359 (3), 357 (2), 332 (3), 331 (4), 319 (5), 294 (4), 289 (2), 287 (2), 274 (3), 262 (2), 257 (3), 241 (4), 240 (3), 228 (4), 227 (5), 221 (3), 220 (2), 57 (2), 43 (2); HRMS (ESI pos, methanol): m/z: calcd for C33H37N2O8 [M + H]+: 589.254992; found: 589.25477.
Methyl 3-{5-[(4-ethyl-3-methyl-5-oxothiophen-2(5H)-ylidene)methyl]-2-formyl-4-methyl-1H-pyrrol-3-yl}propanoate (18): See synthesis of 16. Yellow colored solid (32 mg, 0.09 mmol, 34%). m.p. 151°C; 1H NMR (400 MHz, CDCl3) δ = 1.07 (t, 3J = 7.55 Hz, 3H, 92-CH3), 2.14 (s, 3H, 41-CH3), 2.29 (s, 3H, 81-CH3), 2.44 (q, 3J = 7.52 Hz, 2H, 91-CH2), 2.57 (t, 3J = 7.55 Hz, 2H, 32-CH2), 3.04 (t, 3J = 7.55 Hz, 2H, 31-CH2), 3.65 (s, 3H, CO2CH3), 6.93 (s, 1H, 6-CH), 9.04 (br s, 1H, NH), 9.71 (s, 1H, CHO); 13C NMR (101 MHz, CDCl3) δ = 8.96 (C-41), 12.66 (C-92), 13.06 (C-91), 18.96 (C-81), 19.07 (C-31), 35.20 (C-32), 51.75 (CO2CH3), 113.39 (C-6), 125.31, 131.61, 131.77, 132.40, 134.28 (C-2, -3, -4, -5, -7), 140.77 (C-9), 152.67 (C-8), 172.58 (CO2CH3), 177.65 (CHO), 193.00 (C-10); IR (KBr):  = 3428, 2972, 2934, 2861, 1742, 1673, 1643, 1608, 1584, 1439, 1412, 1385, 1368, 1225, 1203, 1167, 1059, 867, 852, 838, 715, 519, 471 cm−1; UV/Vis [methanol + H2O (5:1)]: λmax = 241, 279, 407 nm (ε not determined); MS (EI, 155°C): m/z (%): 347 (100) [M+], 332 (4), 318 (32), 288 (20), 246 (9), 230 (8), 184 (2), 166 (3), 144 (3), 115 (2), 91 (2), 77 (3), 65 (1), 41 (1); HRMS (ESI pos, methanol + methylene chloride): m/z: calcd for C18H21NNaO4S [M + Na]+: 370.108349; found: 370.108433.
= 3428, 2972, 2934, 2861, 1742, 1673, 1643, 1608, 1584, 1439, 1412, 1385, 1368, 1225, 1203, 1167, 1059, 867, 852, 838, 715, 519, 471 cm−1; UV/Vis [methanol + H2O (5:1)]: λmax = 241, 279, 407 nm (ε not determined); MS (EI, 155°C): m/z (%): 347 (100) [M+], 332 (4), 318 (32), 288 (20), 246 (9), 230 (8), 184 (2), 166 (3), 144 (3), 115 (2), 91 (2), 77 (3), 65 (1), 41 (1); HRMS (ESI pos, methanol + methylene chloride): m/z: calcd for C18H21NNaO4S [M + Na]+: 370.108349; found: 370.108433.
Additionally, the dimerization product 31 (14 mg, 0.022 mmol, 8%) was isolated in the form of a dark blue solid. m.p. 209–211°C; 1H NMR (400 MHz, CDCl3) δ = 1.04 (t, 3J = 7.54 Hz, 6H, 22-, 182-CH3), 2.11 (s, 6H, 71-, 131-CH3), 2.32 (s, 6H, 31-, 171-CH3), 2.39 (q, 3J = 7.52 Hz, 4H, 21-, 181-CH2), 2.55 (t, 3J = 7.55 Hz, 4H, 82-CH2), 2.93 (t, 3J = 7.41 Hz, 4H, 81-CH2), 6.93 (br s, 1H, 10-CH), 7.02 (br s, 2H, 5-, 15-CH); 13C NMR (101 MHz, BB, CDCl3) δ = 11.18 (C-71, -131), 12.76 (C-31, -171), 12.94 (C-22, -182), 18.81 (C21, -181), 19.84 (C-81, -121), 35.10 (C-82, -122), 51.72 (CO2CH3), 115.56 (C-5, -15), 117.26 (C-10), 128.53, 128.79, 130.86, 138.21, 139.31 (C-4, -6, -7, -8, -9, -11, -12, -13, -14, -16), 141.88 (C-2, -18), 152.26 (C-3, -17), 173.03 (CO2CH3), 195.15 (C-1, -19); IR (KBr):  = 3449, 2965, 2931, 2872, 1736, 1666, 1652, 1609, 1458, 1437, 1384, 1364, 1315, 1265, 1197, 1171, 1095, 966, 934, 860 cm−1; UV/Vis (diethyl ether): λmax (ε) = 261 (6700), 328 (13 500), 378 (24 000), 628 nm (13 000); MS (EI, 250°C): m/z (%): 648 (100) [M+], 561 (16), 501 (13), 408 (4), 349 (5), 271 (4), 199 (5), 139 (1), 97 (1), 55 (1).
= 3449, 2965, 2931, 2872, 1736, 1666, 1652, 1609, 1458, 1437, 1384, 1364, 1315, 1265, 1197, 1171, 1095, 966, 934, 860 cm−1; UV/Vis (diethyl ether): λmax (ε) = 261 (6700), 328 (13 500), 378 (24 000), 628 nm (13 000); MS (EI, 250°C): m/z (%): 648 (100) [M+], 561 (16), 501 (13), 408 (4), 349 (5), 271 (4), 199 (5), 139 (1), 97 (1), 55 (1).
Methyl 3-{5-[(3,4-dimethyl-5-oxothiophen-2(5H)-ylidene)methyl]-2-formyl-4-methyl-1H-pyrrol-3-yl}propanoate (19): See synthesis of 16. Yellow colored solid (41 mg, 0.12 mmol, 36%). m.p. 182°C; 1H NMR (400 MHz, CDCl3) δ = 1.98 (s, 3H, 91-CH3), 2.14 (s, 3H, 41-CH3), 2.28 (d, J = 0.80 Hz, 3H, 81-CH3), 2.57 (t, 3J = 7.55 Hz, 2H, 32-CH2), 3.03 (t, 3J = 7.55 Hz, 2H, 31-CH2), 3.66 (s, 3H, CO2CH3), 6.93 (s, 1H, 6-CH), 9.04 (br s, 1H, NH), 9.71 (s, 1H, CHO); 13C NMR (101 MHz, CDCl3) δ = 8.99 (C-41), 10.99 (C-91), 13.01 (C-81), 18.97 (C-31), 35.20 (C-32), 51.77 (CO2CH3), 113.32 (C-6), 125.36, 131.60, 131.76, 132.45, 134.17 (C-2, -3, -4, -5, -7), 135.20 (C-9), 152.96 (C-8), 172.59 (CO2CH3), 177.68 (CHO), 192.11 (C-10); IR (KBr):  = 3426, 2921, 2858, 1727, 1669, 1632, 1609, 1587, 1438, 1417, 1386, 1366, 1317, 1240, 1220, 1167, 1023, 957, 861, 842, 758, 724, 530, 485 cm−1; UV/Vis [methanol + H2O (5:1)]: λmax = 240, 279, 407 nm (ε not determined); MS (EI, 160°C): m/z (%): 333 (100) [M+], 304 (24), 274 (21), 260 (10), 246 (10), 216 (4), 202 (5), 166 (4), 144 (4), 115 (6), 91 (3), 77 (5), 59 (5), 39 (2); HRMS (ESI pos, methanol + methylene chloride): m/z: calcd for C17H19NNaO4S [M + Na]+: 356.092702; found 356.092725.
= 3426, 2921, 2858, 1727, 1669, 1632, 1609, 1587, 1438, 1417, 1386, 1366, 1317, 1240, 1220, 1167, 1023, 957, 861, 842, 758, 724, 530, 485 cm−1; UV/Vis [methanol + H2O (5:1)]: λmax = 240, 279, 407 nm (ε not determined); MS (EI, 160°C): m/z (%): 333 (100) [M+], 304 (24), 274 (21), 260 (10), 246 (10), 216 (4), 202 (5), 166 (4), 144 (4), 115 (6), 91 (3), 77 (5), 59 (5), 39 (2); HRMS (ESI pos, methanol + methylene chloride): m/z: calcd for C17H19NNaO4S [M + Na]+: 356.092702; found 356.092725.
Methyl 3-{5-[(3-ethyl-2-methyl-4-oxocyclopent-2en-1-ylidene)methyl]-2-formyl-4-methyl-1H-pyrrol-3-yl}propanoate (20): See synthesis of 16. Yellow colored spicular crystals (24 mg, 0.07 mmol, 41%). m.p. 186°C; 1H NMR (400 MHz, CDCl3) δ = 1.04 (t, 3J = 7.59 Hz, 3H, 92-CH3), 2.11 (s, 3H, 41-CH3), 2.18 (s, 3H, 81-CH3), 2.34 (q, 3J = 7.57 Hz, 2H, 91-CH2), 2.56 (t, 3J = 7.67 Hz, 2H, 32-CH2), 3.03 (t, 3J = 7.61 Hz, 2H, 31-CH2), 3.20 (s, 2H, 11-CH2), 3.65 (s, 3H, CO2CH3), 6.47 (s, 1H, 6-CH), 8.87 (br s, 1H, NH), 9.63 (s, 1H, CHO); 13C NMR (101 MHz, 1H/13C-COSY, CDCl3) δ = 8.83 (C-41), 11.69 (C-81), 12.90 (C-92), 17.02 (C-91), 19.03 (C-31), 35.34 (C-32), 38.62 (C-11), 51.74 (CO2CH3), 109.31 (C-6), 123.03 (C-4), 130.20 (C-2), 133.03 (C-3), 133.67 (C-5), 136.87 (C-7), 145.77 (C-9), 162.76 (C-8), 172.64 (CO2CH3), 177.00 (CHO), 202.42 (C-10); IR (KBr): 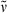 = 3341, 2960, 2934, 2873, 1725, 1677, 1663, 1588, 1552, 1479, 1445, 1395, 1334, 1305, 1273, 1248, 1192, 1168, 1064, 1004, 939, 886, 798, 768, 690, 648, 623 cm−1; UV/Vis [methanol + H2O (5:1)]: λmax = 229, 273, 386 nm (ε not determined); MS (EI, 150°C): m/z (%): 329 (100) [M+], 314 (5), 300 (30), 270 (20), 256 (8), 242 (9), 226 (12), 212 (13), 198 (10), 170 (6), 144 (4), 130 (6), 106 (5), 91 (6), 77 (6), 65 (3), 53 (3), 41 (5); HRMS (ESI pos, methanol + methylene chloride): m/z: calcd for C19H23NNaO4 [M + Na]+: 352.151930; found 352.151556.
= 3341, 2960, 2934, 2873, 1725, 1677, 1663, 1588, 1552, 1479, 1445, 1395, 1334, 1305, 1273, 1248, 1192, 1168, 1064, 1004, 939, 886, 798, 768, 690, 648, 623 cm−1; UV/Vis [methanol + H2O (5:1)]: λmax = 229, 273, 386 nm (ε not determined); MS (EI, 150°C): m/z (%): 329 (100) [M+], 314 (5), 300 (30), 270 (20), 256 (8), 242 (9), 226 (12), 212 (13), 198 (10), 170 (6), 144 (4), 130 (6), 106 (5), 91 (6), 77 (6), 65 (3), 53 (3), 41 (5); HRMS (ESI pos, methanol + methylene chloride): m/z: calcd for C19H23NNaO4 [M + Na]+: 352.151930; found 352.151556.
Methyl 3-{5-[(2,3-dimethyl-4-oxocyclopent-2en-1-ylidene)methyl]-2-formyl-4-methyl-1H-pyrrol-3-yl}propanoate (21): See synthesis of 16. Yellow colored spicular crystals (30 mg, 0.09 mmol, 33%). m.p. 196°C; 1H NMR (400 MHz, CDCl3) δ = 1.87 (s, 3H, 91-CH3), 2.11 (s, 3H, 41-CH3), 2.17 (s, 3H, 81-CH3), 2.56 (t, 3J = 7.60 Hz, 2H, 32-CH2), 3.03 (t, 3J = 7.59 Hz, 2H, 31-CH2), 3.20 (s, 2H, 11-CH2), 3.65 (s, 3H, CO2CH3), 6.47 (s, 1H, 6-CH), 8.84 (br s, 1H, NH), 9.63 (s, 1H, CHO); 13C NMR (101 MHz, 1H/13C-COSY, CDCl3) δ = 8.84 (C-41), 8.88 (C-91), 11.96 (C-81), 19.04 (C-31), 35.31 (C-32), 38.46 (C-11), 51.76 (CO2CH3), 109.05 (C-6), 122.89 (C-4), 129.75 (C-2), 132.35 (C-3), 133.43 (C-5), 136.35 (C-7), 140.05 (C-9), 162.89 (C-8), 172.14 (CO2CH3), 176.27 (CHO), 202.23 (C-10); IR (KBr): 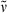 = 3340, 2953, 2917, 2858, 1725, 1682, 1656, 1596, 1550, 1479, 1445, 1395, 1347, 1306, 1262, 1236, 1192, 1167, 1093, 1065, 1012, 938, 881, 783, 690, 657, 625 cm−1; UV/Vis [methanol + H2O (5:1)]: λmax = 277, 386 nm (ε not determined); MS (EI, 160°C): m/z (%): 315 (100) [M+], 300 (2), 286 (30), 256 (24), 242 (10), 228 (10), 212 (14), 198 (9), 184 (8), 170 (6), 144 (3), 130 (4), 106 (3), 91 (3), 77 (3), 65 (1), 55 (1), 41 (1); HRMS (ESI pos, methanol + methylene chloride): m/z: calcd for C18H21NNaO4 [M + Na]+: 338.136279; found 338.136020.
= 3340, 2953, 2917, 2858, 1725, 1682, 1656, 1596, 1550, 1479, 1445, 1395, 1347, 1306, 1262, 1236, 1192, 1167, 1093, 1065, 1012, 938, 881, 783, 690, 657, 625 cm−1; UV/Vis [methanol + H2O (5:1)]: λmax = 277, 386 nm (ε not determined); MS (EI, 160°C): m/z (%): 315 (100) [M+], 300 (2), 286 (30), 256 (24), 242 (10), 228 (10), 212 (14), 198 (9), 184 (8), 170 (6), 144 (3), 130 (4), 106 (3), 91 (3), 77 (3), 65 (1), 55 (1), 41 (1); HRMS (ESI pos, methanol + methylene chloride): m/z: calcd for C18H21NNaO4 [M + Na]+: 338.136279; found 338.136020.
18-Ethyl-17-methyl-oxa-PCB-dimethyl ester (23). The AB building block 22 (18.4 mg, 0.04 mmol), dissolved in TFA (0.9 mL, 11.6 mmol), was stirred for 30 min at ambient temperature in the dark and under argon. After cooling down to −12°C within 5 min a solution of the aldehyde 16 (16.0 mg, 0.05 mmol) in TFA (4.2 mL, 54.0 mmol) was added. The resulting mixture was stirred for 8 h at −12°C. Methanol (1.9 mL, 46.9 mmol) was added and the solution was stirred for additional 30 min at ambient temperature. After evaporation of the solvents in a stream of argon, the remaining solid was dissolved in methylene chloride (30 mL) and washed with a saturated solution of NaHCO3 (30 mL). Afterwards, the organic layer was dried over Na2SO4, the solvent was evaporated in vacuo, and the remaining crude product was purified by HPLC (column A) with methanol and H2O (5:1) as eluent. Evaporation of methanol, followed by extraction of the aqueous dispersion with methylene chloride (3 × 30 mL), drying of the combined organic layers over Na2SO4, and evaporation of the solvent in vacuo yielded the expected reaction product 23 (15.8 mg, 0.03 mmol, 63%) as a dark blue solid. m.p. 189–191°C; 1H NMR (400 MHz, pyridine-d5) δ = 1.06 (t, 3J = 8.61 Hz, 3H, 182-CH3), 1.41 (d, 3J = 7.52 Hz, 3H, 21-CH3), 1.64 (dd, 3Jd = 7.28 Hz, Jd = 0.76 Hz, 3H, 32-CH3), 1.87 (s, 3H, 71-CH3), 1.97 (s, 3H, 171-CH3), 1.99 (s, 3H, 131-CH3), 2.25 (q, 3J = 7.57 Hz, 2H, 181-CH2), 2.62 (t, 3J = 7.42 Hz, 2H, 82-, 122-CH2), 2.66 (t, 3J = 7.53 Hz, 2H, 82-, 122-CH2), 2.96 (t, 3J = 7.44 Hz, 2H, 81-, 121-CH2), 2.98 (t, 3J = 7.42 Hz, 2H, 81-, 121-CH2), 3.22 (q, 3J = 7.63 Hz, 1H, 2-CH), 3.53 (s, 3H, CO2CH3), 3.54 (s, 3H, CO2CH3), 5.94 (s, 1H, 5-CH), 5.99 (s, 1H, 15-CH), 6.31 (dq, Jd = 1.95 Hz, 3Jq = 6.93 Hz, 1H, 31-CH), 7.02 (s, 1H, 10-CH); IR (KBr):  = 2925, 2857, 1737, 1638, 1622, 1592, 1457, 1384, 1315, 1230, 1168, 1129, 1100, 945, 800, 699 cm−1; UV/Vis (methanol): λmax (ε) = 279 (6400), 363 (15 200), 603 nm (4800); MS (EI, 235°C): m/z (%): 615 (50) [M+], 600 (100), 584 (4), 556 (2), 528 (8), 468 (3), 440 (2), 316 (7), 255 (2), 227 (4), 158 (1); MS (ESI pos, methanol): m/z (%): 242 (2), 300 (3), 413 (4), 525 (5), 616 (71) [M + H]+, 638 (100) [M + Na]+, 654 (20) [M + K]+, 700 (27); HRMS (ESI pos, methanol + methylene chloride): m/z: calcd for C35H41N3NaO7 [M + Na]+: 638.283672; found 638.283886.
= 2925, 2857, 1737, 1638, 1622, 1592, 1457, 1384, 1315, 1230, 1168, 1129, 1100, 945, 800, 699 cm−1; UV/Vis (methanol): λmax (ε) = 279 (6400), 363 (15 200), 603 nm (4800); MS (EI, 235°C): m/z (%): 615 (50) [M+], 600 (100), 584 (4), 556 (2), 528 (8), 468 (3), 440 (2), 316 (7), 255 (2), 227 (4), 158 (1); MS (ESI pos, methanol): m/z (%): 242 (2), 300 (3), 413 (4), 525 (5), 616 (71) [M + H]+, 638 (100) [M + Na]+, 654 (20) [M + K]+, 700 (27); HRMS (ESI pos, methanol + methylene chloride): m/z: calcd for C35H41N3NaO7 [M + Na]+: 638.283672; found 638.283886.
17,18-Dimethyl-oxa-PCB-dimethyl ester (24): See synthesis of 23. Dark blue solid (11.5 mg, 0.02 mmol, 71%). m.p. 228°C; 1H NMR (400 MHz, pyridine-d5) δ = 1.47 (d, 3J = 7.51 Hz, 3H, 21-CH3), 1.68 (d, 3J = 7.28 Hz, 3H, 32-CH3), 1.81 (s, 3H, 181-CH3), 1.90 (s, 3H, 71-CH3), 2.02 (s, 3H, 171-CH3), 2.04 (s, 3H, 131-CH3), 2.67 (t, 3J = 7.53 Hz, 2H, 82-, 122-CH2), 2.71 (t, 3J = 7.72 Hz, 2H, 82-, 122-CH2), 3.02 (t, 3J = 6.84 Hz, 4H, 81-, 121-CH2), 3.25 (q, 3J = 7.34 Hz, 1H, 2-CH), 3.58 (s, 3H,CO2CH3), 3.59 (s, 3H, CO2CH3), 5.99 (s, 1H, 5-CH), 6.05 (s, 1H, 15-CH), 6.35 (dq, Jd = 2.26 Hz, 3Jq = 7.21 Hz, 1H, 31-CH), 7.08 (s, 1H, 10-CH); IR (KBr): 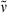 = 2922, 2855, 1740, 1724, 1643, 1622, 1591, 1457, 1411, 1384, 1347, 1314, 1229, 1166, 1129, 1095, 998, 908, 798, 751, 699 cm−1; UV/Vis (methanol): λmax (ε) = 275 (5800), 361 (14 200), 605 nm (4400); MS (EI, 240°C): m/z (%): 601 (49) [M+], 586 (100), 570 (4), 514 (9), 482 (2), 454 (3), 289 (15), 216 (12), 158 (1), 106 (1); MS (ESI pos, methanol): m/z (%): 153 (1), 360 (1), 489 (1), 602 (9) [M + H]+, 624 (100) [M + Na]+, 646 (5), 722 (1); HRMS (ESI pos, methanol + methylene chloride): m/z: calcd for C34H40N3O7 [M + H]+: 602.286079; found 602.285919.
= 2922, 2855, 1740, 1724, 1643, 1622, 1591, 1457, 1411, 1384, 1347, 1314, 1229, 1166, 1129, 1095, 998, 908, 798, 751, 699 cm−1; UV/Vis (methanol): λmax (ε) = 275 (5800), 361 (14 200), 605 nm (4400); MS (EI, 240°C): m/z (%): 601 (49) [M+], 586 (100), 570 (4), 514 (9), 482 (2), 454 (3), 289 (15), 216 (12), 158 (1), 106 (1); MS (ESI pos, methanol): m/z (%): 153 (1), 360 (1), 489 (1), 602 (9) [M + H]+, 624 (100) [M + Na]+, 646 (5), 722 (1); HRMS (ESI pos, methanol + methylene chloride): m/z: calcd for C34H40N3O7 [M + H]+: 602.286079; found 602.285919.
18-Ethyl-17-methyl-thia-PCB-dimethyl ester (25): See synthesis of 23. Purified by HPLC (column D) with a mixture of KH2PO4 buffer (7 mm, pH 7) and acetonitrile (gradient 7:3–1:4) as eluent. Dark blue solid (16.5 mg, 0.03 mmol, 65%). m.p. 143°C; 1H NMR (400 MHz, CDCl3) δ = 1.07 (t, 3J = 7.31 Hz, 3H, 182-CH3), 1.41–1.46 (m, 3H, 21-CH3), 1.69 (d, 3J = 6.48 Hz, 3H, 32-CH3), 1.91–1.95 (m, 3H, 71-CH3), 2.14 (s, 3H, 131-CH3), 2.29 (s, 3H, 171-CH3), 2.44 (q, 3J = 6.40 Hz, 2H, 181-CH2), 2.57 (t, 3J = 7.53 Hz, 4H, 82-, 122-CH2), 3.04 (t, 3J = 7.55 Hz, 4H, 81-, 121-CH2), 3.31–3.36 (m, 1H, 2-CH), 3.63 (d, J = 2.68 Hz, 3H, CO2CH3), 3.65 (d, J = 1.49 Hz, 3H, CO2CH3), 5.87 (s, 1H, 5-CH), 6.43 (s, 1H, 15-CH), 6.51–6.58 (m, 1H, 31-CH), 6.98 (s, 1H, 10-CH); IR (KBr): 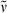 = 3443, 2925, 2855, 1734, 1637, 1560, 1507, 1458, 1438, 1384, 1339, 1261, 1211, 1168, 1130, 1107, 1038, 984, 854, 805, 774, 527, 498 cm−1; UV/Vis (methanol): λmax (ε) = 279 (9800), 379 (13 100), 629 nm (5500); MS (EI, 250°C): m/z (%): 631 (52) [M+], 616 (100), 600 (5), 544 (7), 492 (5), 376 (6), 319 (7), 300 (20), 241 (37), 227 (6), 180 (3), 150 (26), 135 (8), 93 (15), 43 (4); MS (ESI pos, methanol + methylene chloride): m/z (%):370 (16), 632 (61) [M + H]+, 654 (100) [M + Na]+, 1001 (10), 1264 (14) [2 M + H]+, 1286 (27) [2 M + Na]+; HRMS (ESI pos, methanol + methylene chloride): m/z: calcd for C35H42N3O6S [M + H]+: 632.278887; found 632.278375.
= 3443, 2925, 2855, 1734, 1637, 1560, 1507, 1458, 1438, 1384, 1339, 1261, 1211, 1168, 1130, 1107, 1038, 984, 854, 805, 774, 527, 498 cm−1; UV/Vis (methanol): λmax (ε) = 279 (9800), 379 (13 100), 629 nm (5500); MS (EI, 250°C): m/z (%): 631 (52) [M+], 616 (100), 600 (5), 544 (7), 492 (5), 376 (6), 319 (7), 300 (20), 241 (37), 227 (6), 180 (3), 150 (26), 135 (8), 93 (15), 43 (4); MS (ESI pos, methanol + methylene chloride): m/z (%):370 (16), 632 (61) [M + H]+, 654 (100) [M + Na]+, 1001 (10), 1264 (14) [2 M + H]+, 1286 (27) [2 M + Na]+; HRMS (ESI pos, methanol + methylene chloride): m/z: calcd for C35H42N3O6S [M + H]+: 632.278887; found 632.278375.
17,18-Dimethyl-thia-PCB-dimethyl ester (26): See synthesis of 25. Dark blue solid (13.7 mg, 0.02 mmol, 54%). m.p. 188°C; 1H NMR (400 MHz, CDCl3) δ = 1.37 (dd, Jd = 0.91, 3Jd = 6.56 Hz, 3H, 21-CH3), 1.92 (d, 3J = 7.71 Hz, 3H, 32-CH3), 1.94 (s, 3H, 71-CH3), 2.07 (s, 3H, 181-CH3), 2.15 (dd, Jd =2.66 Hz, Jd = 6.87 Hz, 3H, 131-CH3), 2.29 (d, J = 2.32 Hz, 3H, 171-CH3), 2.55 (t, 3J = 8.25 Hz, 4H, 82-, 122-CH2), 2.96 (t, 3J = 7.74 Hz, 4H, 81-, 121-CH2), 3.16–3.23 (m, 1H, 2-CH), 3.64 (s, 3H, CO2CH3), 3.66 (, 3H, CO2CH3), 5.86 (s, 1H, 5-CH), 6.36–6.54 (m, 1H, 31-CH), 5.95 (s, 1H, 15-CH), 6.98 (s, 1H, 10-CH); IR (KBr): 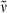 = 3449, 2930, 1734, 1664, 1594, 1523, 1458, 1439, 1384, 1319, 1209, 1168, 1122, 1109, 1039, 931, 864, 806, 683, 636 cm−1; UV/Vis (methanol): λmax (ε) = 279 (7000), 380 (13 300), 595 (3600), 620 nm (3600); MS (EI, 275°C): m/z (%): 617 (62) [M+], 602 (100), 530 (6), 470 (3), 307 (14), 268 (2), 227 (3), 202 (3), 132 (3), 77 (2), 57 (2); HRMS (ESI pos, methanol + methylene chloride): m/z: calcd for C34H40N3O6S [M + H]+: 618.263234; found 618.263043.
= 3449, 2930, 1734, 1664, 1594, 1523, 1458, 1439, 1384, 1319, 1209, 1168, 1122, 1109, 1039, 931, 864, 806, 683, 636 cm−1; UV/Vis (methanol): λmax (ε) = 279 (7000), 380 (13 300), 595 (3600), 620 nm (3600); MS (EI, 275°C): m/z (%): 617 (62) [M+], 602 (100), 530 (6), 470 (3), 307 (14), 268 (2), 227 (3), 202 (3), 132 (3), 77 (2), 57 (2); HRMS (ESI pos, methanol + methylene chloride): m/z: calcd for C34H40N3O6S [M + H]+: 618.263234; found 618.263043.
18-Ethyl-17-methyl-carba-PCB-dimethyl ester (27): See synthesis of 23. Dark blue solid (15.1 mg, 0.03 mmol, 65%). m.p. 198°C; 1H NMR (400 MHz, CDCl3) δ = 1.07 (t, 3J = 7.54 Hz, 3H, 182-CH3), 1.42 (d, 3J = 7.45 Hz, 3H, 21-CH3), 1.93 (d, 3J = 7.56 Hz, 3H, 32-CH3), 2.10 (s, 3H, 71-CH3), 2.13 (s, 3H, 131-CH3), 2.21 (s, 3H, 171-CH3), 2.35 (q, 3J = 7.42 Hz, 2H, 181-CH2), 2.56 (t, 3J = 7.63 Hz, 2H, 82-, 122-CH2), 2.59 (t, 3J = 7.55 Hz, 2H, 82-, 122-CH2), 3.01 (t, 3J = 7.11 Hz, 2H, 81-, 121-CH2), 3.04 (t, 3J = 7.46 Hz, 2H, 81-, 121-CH2), 3.22 (q, 3J = 8.28 Hz, 1H, 2-CH), 3.55 (s, 1H, 20-CH2), 3.59 (s, 1H, 20-CH2), 3.63 (s, 3H, CO2CH3), 3.65 (s, 3H, CO2CH3), 5.28 (s, 1H, 15-CH), 5.86 (s, 1H, 5-CH), 6.50 (dq, Jd = 1.86 Hz, 3Jq = 6.64 Hz, 1H, 31-CH), 6.67 (s, 1H, 10-CH); IR (KBr): 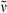 = 2974, 2937, 2872, 1733, 1722, 1687, 1622, 1594, 1452, 1439, 1405, 1384, 1317, 1282, 1244, 1226, 1210, 1190, 1168, 1098, 1057, 966, 938, 832, 746, 690 cm−1; UV/Vis (methanol): λmax (ε) = 267 (8600), 291 (7700), 361 (17 200), 604 nm (7900); MS (EI, 240°C): m/z (%): 613 (79) [M+], 598 (100), 526 (9), 492 (2), 466 (3), 300 (6), 254 (2), 226 (1), 55 (1); MS (ESI pos, methanol): m/z (%): 304 (2), 367 (2), 457 (2), 529 (2), 614 (20) [M + H]+, 636 (100) [M + Na]+, 652 (15) [M + K]+, 675 (68), 766 (4); HRMS (ESI pos, methanol + methylene chloride): m/z: calcd for C36H43N3NaO6 [M + Na]+: 636.304403; found 636.303838.
= 2974, 2937, 2872, 1733, 1722, 1687, 1622, 1594, 1452, 1439, 1405, 1384, 1317, 1282, 1244, 1226, 1210, 1190, 1168, 1098, 1057, 966, 938, 832, 746, 690 cm−1; UV/Vis (methanol): λmax (ε) = 267 (8600), 291 (7700), 361 (17 200), 604 nm (7900); MS (EI, 240°C): m/z (%): 613 (79) [M+], 598 (100), 526 (9), 492 (2), 466 (3), 300 (6), 254 (2), 226 (1), 55 (1); MS (ESI pos, methanol): m/z (%): 304 (2), 367 (2), 457 (2), 529 (2), 614 (20) [M + H]+, 636 (100) [M + Na]+, 652 (15) [M + K]+, 675 (68), 766 (4); HRMS (ESI pos, methanol + methylene chloride): m/z: calcd for C36H43N3NaO6 [M + Na]+: 636.304403; found 636.303838.
17,18-Dimethyl-carba-PCB-dimethyl ester (28): See synthesis of 23. Dark blue solid (11.8 mg, 0.02 mmol, 48%). m.p. 196°C; 1H NMR (400 MHz, 1H/1H-COSY, pyridine-d5) δ = 1.48 (d, 3J = 7.54 Hz, 3H, 21-CH3), 1.70 (d, 3J = 7.28 Hz, 3H, 32-CH3), 1.78 (s, 3H, 181-CH3), 1.99 (s, 3H, 71-CH3), 2.11 (s, 6H, 131-, 171-CH3), 2.72 (t, 3J = 7.13 Hz, 2H, 82-, 122-CH2), 2.75 (t, 3J = 7.03 Hz, 2H, 82-, 122-CH2), 3.10 (t, 3J = 7.33 Hz, 4H, 81-, 121-CH2), 3.44–3.47 (m, 1H, 2-CH), 3.48 (s, 1H, 20-CH2), 3.59 (s, 3H, CO2CH3), 3.60 (s, 3H, CO2CH3), 3.63 (s, 1H, 20-CH2), 6.08 (s, 1H, 5-CH), 6.36 (q, 3J = 6.86 Hz, 1H, 31-CH), 6.55 (s, 1H, 15-CH), 7.13 (s, 1H, 10-CH); IR (KBr): 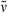 = 2923, 2855, 1728, 1687, 1621, 1589, 1441, 1406, 1384, 1317, 1213, 1169, 1089, 1056, 1041, 965, 879, 837, 808, 746, 693, 651, 617, 441 cm−1; UV/Vis (methanol): λmax (ε) = 266 (8200), 293 (7300), 362 (16 800), 603 nm (7900); MS (EI, 240°C): m/z (%): 601 (13), 600 (36), 599 (92) [M+], 586 (9), 585 (38), 584 (100), 512 (9), 452 (5), 300 (19), 214 (6), 180 (3), 105 (6); MS (ESI pos, methanol + methylene chloride): m/z (%): 600 (22) [M + H]+, 601 (8), 621 (3), 622 (100) [M + Na]+, 623 (38), 624 (8), 644 (7), 720 (4), 1199 (21) [2M + H]+, 1200 (15), 1201 (62), 1221 (58) [2M + Na]+, 1222 (59), 1223 (18); HRMS (ESI pos, methanol + methylene chloride): m/z: calcd for C35H41N3NaO6 [M + Na]+: 622.288753; found 622.288616.
= 2923, 2855, 1728, 1687, 1621, 1589, 1441, 1406, 1384, 1317, 1213, 1169, 1089, 1056, 1041, 965, 879, 837, 808, 746, 693, 651, 617, 441 cm−1; UV/Vis (methanol): λmax (ε) = 266 (8200), 293 (7300), 362 (16 800), 603 nm (7900); MS (EI, 240°C): m/z (%): 601 (13), 600 (36), 599 (92) [M+], 586 (9), 585 (38), 584 (100), 512 (9), 452 (5), 300 (19), 214 (6), 180 (3), 105 (6); MS (ESI pos, methanol + methylene chloride): m/z (%): 600 (22) [M + H]+, 601 (8), 621 (3), 622 (100) [M + Na]+, 623 (38), 624 (8), 644 (7), 720 (4), 1199 (21) [2M + H]+, 1200 (15), 1201 (62), 1221 (58) [2M + Na]+, 1222 (59), 1223 (18); HRMS (ESI pos, methanol + methylene chloride): m/z: calcd for C35H41N3NaO6 [M + Na]+: 622.288753; found 622.288616.
18-Ethyl-17-methyl-oxa-PCB (4). The ester 23 (11.1 mg, 0.02 mmol) was dissolved in a mixture of TFA and H2O (30 mL, 1:1, vol/vol). Subsequently, acidic ion exchange resin DOWEX 50W×8−200 (12.4 g) which was washed three times with H2O and dried in a stream of air before it was added and the dispersion was stirred for 48 h at ambient temperature in the dark. The ion exchange resin was filtered off (sintered glass filter, size 4) and washed alternately with H2O (5 × 40 mL) and a solution of chloroform and methanol (49:1, 5 × 40 mL). The aqueous layer of the combined filtrates was separated and extracted with a solution of chloroform and methanol (49:1, 4 × 40 mL). Afterwards the combined organic phases were washed with brine (50 mL portions) until pH 6 and dried over Na2SO4. Evaporation of the solvent in vacuo yielded the crude product (17.6 mg) which was purified by HPLC (column B) with a mixture of ammonium acetate buffer (10 mm, pH 7) and methanol (gradient 1:1–1:8) as eluent. Evaporation of the methanol followed by extraction of the aqueous dispersion with methylene chloride (5 × 30 mL), drying of the combined organic layers over Na2SO4, and evaporation of the solvent in vacuo yielded the expected reaction product 4 (7.8 mg, 0.01 mmol, 74%) as a dark blue glowing solid. Decomposition until 335°C; 1H NMR (400 MHz, 1H/1H-COSY, pyridine-d5) δ = 1.11 (t, 3J = 7.55 Hz, 3H, 182-CH3), 1.46 (d, 3J = 7.52 Hz, 3H, 21-CH3), 1.68 (dd, 3Jd = 7.35 Hz, Jd = 0.64 Hz, 3H, 32-CH3), 1.91 (s, 3H, 71-CH3), 2.06 (s, 3H, 171-CH3), 2.07 (s, 3H, 131-CH3), 2.29 (q, 3J = 7.45 Hz, 2H, 181-CH2), 2.84 (t, 3J = 7.35 Hz, 2H, 82-, 122-CH2), 2.89 (t, 3J = 7.54 Hz, 2H, 82-, 122-CH2), 3.15 (t, 3J = 7.32 Hz, 2H, 81-, 121-CH2), 3.15 (t, 3J = 7.21 Hz, 2H, 81-, 121-CH2), 3.26 (dq, Jd = 0.65 Hz, 3Jq = 7.52 Hz, 1H, 2-CH), 5.97 (s, 1H, 5-CH), 6.02 (s, 1H, 15-CH), 6.33 (dq, Jd = 2.31 Hz, 3Jq = 7.33 Hz, 1H, 31-CH), 7.30 (s, 1H, 10-CH); UV/Vis [methanol + H2O (3:1)]: λmax = 279, 367, 611 nm (ε not determined); MS (ESI pos, methanol + methylene chloride): m/z (%): 441 (28), 491 (100), 553 (22), 588 (59) [M + H]+, 589 (20), 590 (5), 610 (37) [M + Na]+, 611 (15), 632 (18), 654 (19); MS (MALDI-TOF pos, α-CHCA): m/z (%): 573 (19) [M – 15]+, 574 (41) [M – 14]+, 575 (19), 576 (5), 587 (22), 588 (82) [M+], 589 (100) [M + H]+, 590 (91), 610 (9), 611 (35) [M + Na]+, 612 (16), 613 (6), 627 (6) [M + K]+; HRMS (ESI pos, methanol + methylene chloride): m/z: calcd for C33H38N3O7 [M + H]+: 588.270429; found 588.271108.
17,18-Dimethyl-oxa-PCB (5): See synthesis of 4. Dark blue solid (13.3 mg, 0.02 mmol, 70%). Decomposition until 335°C; 1H NMR (400 MHz, 1H/1H-COSY, pyridine-d5) δ = 1.47 (d, 3J = 7.21 Hz, 3H, 21-CH3), 1.68 (d, 3J = 7.21 Hz, 3H, 32-CH3), 1.81 (s, 3H, 181-CH3), 1.89 (s, 3H, 71-CH3), 2.06 (s, 3H, 171-CH3), 2.07 (s, 3H, 131-CH3), 2.85 (t, 3J = 7.28 Hz, 2H, 82-, 122-CH2), 2.89 (t, 3J = 7.43 Hz, 2H, 82-, 122-CH2), 3.15 (t, 3J = 6.87 Hz, 2H, 81-, 121-CH2), 3.16 (t, 3J = 7.50 Hz, 2H, 81-, 121-CH2), 3.24 (q, 3J = 7.74 Hz, 1H, 2-CH), 5.98 (s, 1H, 5-CH), 6.04 (s, 1H, 15-CH), 6.35 (dq, Jd = 1.82 Hz, 3Jq = 6.88 Hz, 1H, 31-CH), 7.32 (s, 1H, 10-CH); UV/Vis [methanol + H2O (3:1)]: λmax = 279, 366, 613 nm (ε not determined); MS (ESI pos, methanol): m/z (%): 357 (10), 393 (10), 413 (25), 489 (11), 574 (100) [M + H]+, 575 (37), 576 (10), 577 (5), 596 (73) [M + Na]+, 597 (30), 598 (6), 599 (5), 618 (33), 640 (27); MS (ESI neg, methanol): m/z (%): 468 (11), 528 (42), 529 (15), 572 (83) [M − H]−, 573 (31), 574 (7), 575 (2), 594 (100), 595 (38), 596 (7), 597 (3), 758 (3), 813 (47), 814 (24); MS (MALDI-TOF pos, α-CHCA): m/z (%): 523 (11), 524 (5), 551 (12), 552 (6), 559 (4) [M − 15]+, 560 (15) [M − 14]+, 561 (7), 573 (10), 574 (78) [M]+, 575 (100) [M + H]+, 576 (68), 577 (21), 597 (5) [M + Na]+, 612 (3), 613 (10) [M + K]+ 614 (4); HRMS (ESI pos, methanol + methylene chloride): m/z: calcd for C32H36N3O7 [M + H]+: 574.254778; found 574.254557.
18-Ethyl-17-methyl-thia-PCB (6): See synthesis of 4. Purified by HPLC (column B) with a mixture of KH2PO4 buffer (7 mm, pH 7) and acetonitrile (gradient 7:3–1:4) as eluent. Dark blue solid (16.9 mg, 0.03 mmol, 69%). Decomposition until 335°C; 1H NMR (400 MHz, pyridine-d5) δ = 1.06–1.10 (m, 3H, 182-CH3), 1.49 (d, 3J = 7.48 Hz, 3H, 21-CH3), 1.69 (d, 3J = 7.09 Hz, 3H, 32-CH3), 1.94 (s, 3H, 71-CH3), 2.14 (s, 3H, 171-CH3), 2.15 (d, J = 2.27 Hz, 3H, 131-CH3), 2.41 (q, 3J = 7.47 Hz, 2H, 181-CH2), 2.87 (t, 3J = 7.26 Hz, 4H, 82-, 122-CH2), 3.18 (t, 3J = 7.58 Hz, 2H, 81-, 121-CH2), 3.20 (t, 3J = 7.52 Hz, 2H, 81-, 121-CH2), 3.36 (q, 3J = 7.97 Hz, 1H, 2-CH), 5.67 (s, 1H, 5-CH), 6.05 (s, 1H, 15-CH), 6.35 (q, 3J = 6.85 Hz, 1H, 31-CH), 7.24 (s, 1H, 10-CH); UV/Vis [acetonitrile + H2O (1:1)]: λmax = 271, 294, 382, 609, 630 nm (ε not determined); MS (ESI neg, methanol): m/z (%): 147 (4), 255 (4), 339 (5), 397 (1), 513 (1), 558 (6), 602 (100) [M − H]−, 663 (7), 723 (3); MS (MALDI-TOF pos, α-CHCA): m/z (%): 589 (2) [M − 15]+, 590 (12) [M − 14]+, 591 (5), 603 (6), 604 (35) [M+], 605 (100) [M + H]+, 606 (55), 607 (27), 608 (9), 626 (4), 627 (15) [M + Na]+, 628 (10); HRMS (EI): m/z: calcd for C33H38N3O6S [M + H]+: 604.247585; found 604.247875.
17,18-Dimethyl-thia-PCB (7): See synthesis of 6. Dark blue solid (12.0 mg, 0.02 mmol, 92%). Decomposition until 335°C; 1H NMR (500 MHz, 1H/1H-COSY, pyridine-d5) δ = 1.48 (d, 3J = 7.54 Hz, 3H, 21-CH3), 1.69 (d, 3J = 7.26 Hz, 3H, 32-CH3), 1.88 (s, 3H, 71-CH3), 2.11 (s, 3H, 181-CH3), 2.14 (s, 3H, 171-CH3), 2.14 (s, 3H, 131-CH3), 2.87 (t, 3J = 7.29 Hz, 2H, 82-, 122-CH2), 2.91 (t, 3J = 7.50 Hz, 2H, 82-, 122-CH2), 3.18 (t, 3J = 7.27 Hz, 2H, 81-, 121-CH2), 3.20 (t, 3J = 7.38 Hz, 2H, 81-, 121-CH2), 3.37 (q, 3J = 7.57 Hz, 1H, 2-CH), 5.66 (s, 1H, 5-CH), 6.06 (s, 1H, 15-CH), 6.35 (dq, Jd = 2.03 Hz, 3Jq = 7.12 Hz, 1H, 31-CH), 7.23 (s, 1H, 10-CH); UV/Vis [acetonitrile + H2O (1:1)]: λmax = 271, 294, 382, 609, 629 nm (ε not determined); UV/Vis [H2O + methanol (4:1)]: λmax = 294, 382, 629 nm (ε not determined); MS (ESI neg, methanol): m/z (%): 265 (3), 339 (18), 397 (1), 441 (1), 485 (1), 544 (9), 588 (100) [M − H]–, 649 (3), 708 (1); MS (MALDI-TOF pos, α-CHCA): m/z (%): 575 (5) [M − 15]+, 576 (9) [M − 14]+, 578 (5), 589 (12), 590 (33) [M+], 591 (100) [M + H]+, 592 (34), 593 (7), 613 (4) [M + Na]+, 614 (4); HRMS (ESI pos, methanol + methylene chloride): m/z: calcd for C32H35N3NaO6S [M + Na]+: 612.213878; found 612.213153.
18-Ethyl-17-methyl-carba-PCB (8): See synthesis of 4. Dark blue solid (7.3 mg, 0.01 mmol, 51%). Decomposition until 335°C; 1H NMR (400 MHz, 1H/1H-COSY, pyridine-d5) δ = 1.04 (t, 3J = 7.53 Hz, 3H, 182-CH3), 1.49 (d, 3J = 7.54 Hz, 3H, 21-CH3), 1.70 (d, 3J = 7.20 Hz, 3H, 32-CH3), 2.02 (s, 3H, 71-CH3), 2.15 (s, 3H, 171-CH3), 2.15 (s, 3H, 131-CH3), 2.32 (dq, Jd = 1.78 Hz, 3Jq = 7.56 Hz, 2H, 181-CH2), 2.89 (t, 3J = 7.32 Hz, 2H, 82-, 122-CH2), 2.93 (t, 3J = 7.58 Hz, 2H, 82-, 122-CH2), 3.21 (t, 3J = 6.27 Hz, 2H, 81-, 121-CH2), 3.23 (t, 3J = 6.35 Hz, 2H, 81-, 121-CH2), 3.44–3.47 (m, 1H, 2-CH), 3.49 (s, 1H, 20-CH2), 3.56 (s, 1H, 20-CH2), 6.06 (s, 1H, 5-CH), 6.34 (dq, Jd = 2.22 Hz, 3Jq = 7.33 Hz, 1H, 31-CH), 6.54 (s, 1H, 15-CH), 7.23 (s, 1H, 10-CH); UV/Vis [methanol + H2O (7:2)]: λmax = 268, 294, 366, 608 nm (ε not determined); MS (ESI pos, methanol + methylene chloride): m/z (%): 586 (100) [M + H]+, 587 (35), 588 (8), 608 (65) [M + Na]+, 609 (25), 610 (5); MS (MALDI-TOF pos, α-CHCA): m/z (%): 571 (4) [M − 15]+, 572 (11) [M − 14]+, 573 (5), 585 (11), 586 (55) M+, 587 (100) [M + H]+, 588 (52), 589 (16), 590 (5), 608 (6), 609 (23) [M + Na]+, 610 (9), 625 (5) [M + K]+, 631 (7), 651 (4), 657 (4); HRMS (ESI pos, methanol + methylene chloride): m/z: calcd for C34H40N3O6 [M + H]+: 586.291161; found 586.291504.
17,18-Dimethyl-carba-PCB (9): See synthesis of 4. Purified by HPLC (column B + C) with a mixture of ammonium acetate buffer (10 mm, pH 7) and methanol (gradient 1:1–1:8) as eluent. Dark blue solid (6.1 mg, 0.01 mmol, 66%). Decomposition until 335°C; 1H NMR (400 MHz, 1H/1H-COSY, pyridine-d5) δ = 1.48 (d, 3J = 7.54 Hz, 3H, 21-CH3), 1.69 (dd, Jd = 0.90 Hz, 3Jd = 7.05 Hz, 3H, 32-CH3), 1.78 (s, 3H, 181-CH3), 1.99 (s, 3H, 71-CH3), 2.14 (s, 3H, 171-CH3), 2.15 (s, 3H, 131-CH3), 2.89 (t, 3J = 7.18 Hz, 2H, 82-, 122-CH2), 2.94 (t, 3J = 7.54 Hz, 2H, 82-, 122-CH2), 3.22 (t, 3J = 6.98 Hz, 2H, 81-, 121-CH2), 3.23 (t, 3J = 7.32 Hz, 2H, 81-, 121-CH2), 3.44–3.48 (m, 1H, 2-CH), 3.50 (s, 1H, 20-CH2), 3.60 (s, 1H, 20-CH2), 6.07 (s, 1H, 5-CH), 6.33 (dq, Jd = 2.50 Hz, 3Jq = 7.34 Hz, 1H, 31-CH), 6.54 (s, 1H, 15-CH), 7.29 (s, 1H, 10-CH); UV/Vis [methanol + H2O (4:1)]: λmax = 268, 294, 365, 606 nm (ε not determined); MS (ESI pos, methanol): m/z (%): 242 (5), 572 (100) [M + H]+, 573 (36), 574 (7), 594 (6), 1143 (18) [2M + H]+, 1144 (13), 1145 (5); MS (ESI neg, methanol): m/z (%):284 (4), 339 (2), 570 (100) [M − H]−, 571 (22), 572 (7), 573 (1), 592 (1), 606 (1); MS (MALDI-TOF pos, α-CHCA): m/z (%): 523 (3), 558 (7) [M − 14]+, 559 (4), 571 (5), 572 (64) [M+], 573 (100) [M + H]+, 574 (56), 575 (14), 595 (3) [M + Na]+, 612 (4); HRMS (ESI pos, methanol + methylene chloride): m/z: calcd for C33H38N3O6[M + H]+: 572.275511; found 572.275946.
Results
Synthesis of novel oxa-, thia- and carba-“bilins” 4–9
A recently reported synthetic approach yielded six pyrryl-furanones, -thiophenones and -cyclopentenones (10–15, Fig. 2) (5) which represent the “right halves” of the convergent bilin synthesis, introduced by Gossauer et al. (7). These compounds were always generated as pairs in 9-ethyl-8-methyl and in 8,9-dimethyl substituted form (the substitution pattern referring to positions 17 and 18 of ring D in the native chromophore, Fig. 1). Systematic improvement of the condensation with the “left half” of PCB as well as of the workup procedure, with respect to the recently published method (8), yielded six PCB-analog compounds, each carrying an oxygen or a sulfur atom, or a methylene group at the position of the pyrrole nitrogen atom in ring D, still in form of their methyl propionates 23–28 (Fig. 3). The coupling reaction takes place in two steps (indicated as a and b in Fig. 3) such that an 8 h treatment of 22 with TFA leads to the removal of its t-butyl ester and a decarboxylation of both free carboxyl functions, followed by formation of the central carbon–carbon bond during reaction with the compounds 16–21. Subsequent addition of methanol allows the generation of the double bond. Saponification employing an ion exchange resin (9) then yielded the final analog chromophores 4–9 in a futher improved yield compared with the literature. Besides the target compounds, dimers of 10–12 formed unintended via their formylation (introduction of a formyl group, finally serving as the central methine bridge is a necessary intermediate step in bilin synthesis, see Fig. 2). These side products (29–31) were easily separated from the target compounds by chromatography and chemically characterized. Although they showed absorbance spectra akin to tetrapyrroles (Fig. 4), due to the small quantity they were not employed for any further studies. The absorbance spectra of compounds 4–9 showed the bilin-typical features such that the most intense absorption was between 363 and 382 nm, and a broad absorption band was found around 620 nm (see Fig. 5 and Table 1; only the spectra of the 18-ethyl-17-methyl compounds are shown, their dimethyl analogs absorb virtually identical). This long-wavelength band usually consists of two closely lying absorption bands with one or the other being of slightly higher intensity. The long-wavelength absorbance maxima of the six novel “PCB-analogs”4–9, together with those of PCB (2) and 17,18-dimethyl PCB (3) (10) can be arranged into two groups: The carba- and the oxa-derivatives 4–7 showed relatively blueshifted absorption maxima of 606 and 608 nm (9, 8), and of 611 and 613 nm (4, 5), whereas the thia- (6, 7) and aza-derivatives PCB (2) and 17,18-dimethyl PCB (3) show absorption maxima with a redshift of 20 and more nm compared with the former ones: 629, 630 nm for 7 and 6, respectively, and 627 and 630 nm for PCB (2) itself and for 17,18-dimethyl PCB (3) (see Table 1).

Coupling reactions between the so-called “left half”22 and the “right halves”16–21 to PCB-like diesters 23–28 and subsequent de-esterification of the propionic acid ester side chains. Yields are given over both reaction-steps.

Kekulé formula and UV/Vis absorption parameters for the dimerization products 29–31 (all values were determined in diethyl ether).

UV/Vis absorption spectra of PCB 2 and the selected analogs 4, 6 and 8 recorded in methanol/H2O (3:1–4:1, vol/vol).
| λ max [nm] (ε [L mol−1 cm−1]) | ||||
|---|---|---|---|---|
| 23 | – | 279 (6400) | 363 (15 200) | 603 (4800) |
| 24 | – | 275 (5800) | 361 (14 200) | 605 (4400) |
| 25 | – | 279 (9800) | 379 (13 100) | 629 (5500) |
| 26 | – | 279 (7000) | 380 (13 300) | 620 (3600) |
| 27 | 267 (8600) | 291 (7700) | 361 (17 200) | 604 (7900) |
| 28 | 266 (8200) | 293 (7300) | 362 (16 800) | 603 (7900) |
| 2 | 270 | 279 | 363 | 603/627 |
| 3 | – | n.d. | 363 | 587/630 |
| 4 | – | 279 | 367 | 611 |
| 5 | – | 279 | 366 | 613 |
| 6 | 271 | 294 | 382 | 630 |
| 7 | 271 | 294 | 382 | 629 |
| 8 | 268 | 294 | 366 | 608 |
| 9 | 268 | 294 | 365 | 606 |
- For reasons of limited material availability, extinction coefficients were not determined for the free acids. n.d., not determined.
Assembly of 4–9 with recombinant apophytochrome
All six analog chromophores formed stable chromoproteins, when incubated with the N-terminal 65 kDa domain of recombinant oat apophytochrome. The absorption spectra of their Pr-forms are much akin that of native phytochrome in showing an absorbance around 350–380 nm of lower intensity and a strong absorbance band in the long wavelength region of the spectrum (see Table 2, Fig. 6; note that the traces in the upper panel show also the spectra of the Pfr-forms after irradiation. In some cases, the changes between both forms are practically negligible, yielding nearly identical, coinciding absorption spectra.). The ratio between the absorbances in the 350–380 nm region and the Pr-peaks was not significantly different to PCB-assembled phytochrome (spectra not shown). Like for phytochrome, the absorbance bands showed a shoulder at the high energy part. This shoulder in the absorption band is most intense for the oxa-derivatives, whereas for the other four compounds, the thia- and the carba-derivatives, it is barely visible. For all six novel chromophores, the absorption bands of the Pr-forms are blueshifted compared with phytochrome or PCB-assembled phytochrome. Still, the binding with the apoprotein causes a bathochromic shift of the absorption of the free chromophores 2–5, 8, 9, except of the thia-derivatives 6, 7, which exhibit practically identical absorption maxima in free- and protein-bound form. Addition of PCB (2) to the novel phytochromes did not change their spectra nor their photochemical reactivity (see below), indicating that the hetero atom-substituted compounds 4–9 occupied the same binding pocket and formed stable covalent bonds to the canonical chromophore-binding cysteine residue of the apophytochrome. The spectral contributions from the analog-assembled phytochromes (structured, phytochrome-typical absorption bands) and from added PCB (2) (broad, unstructured absorption band) were clearly discernible. The absorption from unbound PCB (2) was lost after a gel filtration step, which left the analog-compound absorption bands undisturbed, giving further evidence for the covalently bound PCB-analogs.
| Pr max [nm] | Pfr max [nm] | Δ [cm−1] | λ ex [nm] | λ em [nm] | ΔSt [cm−1] | φ [× 10−3] | |
|---|---|---|---|---|---|---|---|
| 2 | 651 | 711 | 1296 | 656 | 675 | 429 | 3.7 ± 0.4* |
| 3 | 655 | 714 | 1262 | n.d. | n.d. | n.d. | n.d. |
| 4 | 634 | 669 | 825 | 638 | 655 | 407 | 11.3 ± 0.8 |
| 5 | 635 | 670 | 823 | 638 | 653 | 360 | 8.6 ± 1.0 |
| 6 | 629 | 690 | 1405 | 632 | 656 | 579 | 14.2 ± 1.6 |
| 7 | 633 | 690 | 1305 | 637 | 660 | 547 | 9.3 ± 1.1 |
| 8 | 629 | 637† | 199 | 622 | 648 | 645 | 11.4 ± 1.2 |
| 9 | 626 | 641† | 373 | 628 | 647 | 468 | 12.4 ± 1.4 |
- λ ex, excitation wavelength; λex, emission wavelength; ΔSt, Stokes’ shift (λem [cm−1]–λex [cm−1]); n.d., not determined. *Reference (16); †Due to the low photochemical activity, these values are estimated, see text.

Details of the UV/Vis absorption spectra of selected chromoproteins, assembled with 4 (dark gray), 6 (black) and 8 (light gray). Whereas the upper half shows the absorption of the Pr- (continuous lines) and Pfr-states (dotted lines) between 450 and 850 nm, the lower half shows the consequential Pr-Pfr difference spectra. Due to the low photochemical activity of the chromoproteins, the Pr- and Pfr-spectra at some positions coincide. The midpoint wavelengths of the interference filters used for generation of the Pfr-states are indicated by arrows. In the lower panel, the zero-line was introduced to allow an estimation of the spectral changes in particular for the carba-analogs. For sample handling conditions, see Materials and Methods section.
Steady state spectroscopy
Irradiation of phytochromes carrying chromophores 4–9 in the range of their long-wavelength Pr-band (616, 578 and 578 nm, respectively) revealed a very reduced photochemical reactivity. A Pfr-form as well as a difference spectrum between Pr and Pfr could be generated for the oxa- and the thia-derivatives, whereas the carba-compounds showed an extremely diminished photochemical reactivity making a determination of the Pr- and Pfr-maxima uncertain (Fig. 6). The hypsochromic shift of the Pr-maxima at ca. 20 nm, compared with PCB-assembled phytochrome (see previous paragraph), is also found for the Pfr-peaks. However, whereas the Pr-forms absorb in a fairly narrow spectral range, a discrimination is seen for the Pfr-forms: the longest absorption maximum is found for both thia-derivatives (690 nm), the oxa-derivatives generate Pfr-forms at 669 and 670 nm, respectively, and the most blueshifted Pfr-absorption was found for the carba-analogs at 637 and 641 nm, respectively (see Table 2, Fig. 6; the Pfr absorption maxima have been estimated as the centers of very shallow broad bands with 635–640 nm for 8 and 636–645 nm for 9).
Steady state fluorescence measurements showed an at least three-fold higher fluorescence quantum yield for any of the six novel chromoproteins (Table 2); however, the Stokes shifts were of similar size, ranging from 15 nm (360 cm−1) in compound 5-assembled phytochrome up to 26 nm (645 cm−1) in 8-assembled phytochrome (Fig. 7). The higher fluorescence quantum yield for the PCB analogs might be explained by a less intimate interaction with the protein, compared with PCB (2). This native chromophore is apparently optimally incorporated into the protein cavity, allowing a very efficient deactivation of the excited state by interactions with the protein surrounding.

Normalized fluorescence-spectra of phytochromes, assembled with PCB (2) (black) and its derivatives 4 (red), 7 (green) and 8 (blue). Whereas the dashed lines represent the absorption spectra the continuous lines describe the emission spectra. For sample handling conditions, see Materials and Methods section.
Discussion
The light-induced switching between two thermally stable states (Pr and Pfr) is essential for the function of phytochromes. For the Pr-state, the three-dimensional structure of the chromophore-binding domain of D. radiodurans has revealed many precise interactions between chromophore and protein that stabilize the chromophore conformation in its dark state and direct the photochemical activity towards the CD ring region. An explanation of how the thermal stability of the Pfr-state is accomplished, however, remains still elusive, in particular, as the protein cavity around ring D is relatively unfunctionalized and is composed mostly of hydrophobic amino acid side chains. In addition, part of the chromophore binding site might be missing in the crystallized phytochrome domains, as the construct used for these studies consisted only of the PAS- and the GAF-domain. Yet, the crystal structure gives information on how the selectivity of double bond isomerization is directed towards the CD methine bridge. The rings A (by covalent attachment), B and C (by strong interactions of the propionate side chains with the protein) are so tightly fixed that only the CD methine bridge is allowed to isomerize. In particular the role of the propionate groups in dissociated state has been documented by employing a series of chemically synthesized, structurally modified tetrapyrroles (11), and also by the use of mono methyl esters of PCB (2) (12). In addition, the crystal structure of the chromophore-binding domain of the D. radiodurans phytochrome (13) revealed the strong hydrogen-bonding interaction between the protonated ring A, B and C nitrogens with the backbone carbonyl group of Asp207. A modification of the chromophore structure, as already documented by e.g. Inomata et al. (14), thus appeared as a valuable tool to study in greater detail the mechanisms which stabilize the bilin moiety in its photoisomerized Pfr-state. As it has already been shown (10,15), neither a change of the methyl substituent at position 17 nor of the vinyl- (PΦB, 1) or the ethyl-substituent (PCB, 2) at position 18 cause severe changes in the photochemistry of these modified phytochromes, except of a steric hindrance induced by an isopropyl group at position 17 (10). Besides interactions derived from the carbonyl function, which have not been addressed so far, the polar pyrrole ring with its hydrogen donor capacity might contribute to a stabilization of the Pfr-form of phytochrome.
In fact, the variation in polarity outlined here points to an involvement of the nitrogen atom and the attached proton of ring D within the chromophore-protein interactions. The situation is different in the nonprotein bound form: the spectral properties of all six novel compounds 4–7 are very similar to those of PCB (2), except of the fact that in free form the sulfur atom (compounds 6 and 7) and the nitrogen atom PCB (2), due to their greater polarizability, show a larger auxochromic effect than the oxygen atom (4 and 5) or the methylene group (8 and 9). This effect is leveled off in the protein-bound form, where, apparently, other factors govern the position of the Pr-absorption band. Here, the most redshifted absorption of the Pr-form is that of PCB-assembled phytochrome, indicating the importance of hydrogen bonds for the spectral properties of phytochromes. The difference in chemical reactivity becomes even more evident, when the photochemical properties were investigated. None of the three motifs, oxa-, thia- or carba-derivative, underwent a noticeable Pr-to-Pfr conversion, in fact, the diminished Pfr-formation of the carba-compounds 8 and 9 made a detection of the Pfr-maximum nearly impossible. The low photochemical reactivity does not concur with the wavelength difference between the Pr- and the Pfr-form. Although showing the most redshifted absorption maxima (Pr and Pfr), PCB-assembled phytochrome does not undergo the largest bathochromic shift upon Pfr-formation. The difference (in wavenumbers) between both forms of PCB-assembled phytochrome is 1296 and 1262 cm−1 for the 17,18-dimethyl PCB derivative 3. Also the native chromophore PΦB (1) causes a similar Pr-Pfr shift of 1302 cm−1. An even larger difference between the Pr- and Pfr-state is measured for both thia-substituted chromophores: 1405 cm−1 for 6 and 1305 cm−1 for its 17,18-dimethyl derivative 7. On the other hand, the oxa-substituted chromophores generate a significantly smaller shift: 825 and 823 cm−1 for 4 and 5, respectively, and, although most restricted due to the uncertainty of detection, the carba-derivatives 8 and 9 show bathochromic shifts of only 199 and 373 cm−1. Unfortunately, the low quantum yield of these phytochrome analogs impeded a time-resolved study of light-induced reactions which might have given information whether at all an isomerization took place which immediately reverted into the parent Pr-state.
Taken together, the results obtained with the hetero atom derivatives 4–9 of PCB (2) point to a strong contribution of the polarizable nitrogen atom in ring D of the chromophore. All six derivatives (4–9) show a lower polarity of the ring D than PCB (2), and no acidic hydrogen atom is present, giving a strong argument that the conformation of the chromophore in the Pfr-state is stabilized—amongst other forces—by hydrogen bonds and polar interactions between the nitrogen atom and its protein environment.
Acknowledgements— The excellent technical support by Manuela Trinoga in the HPLC purifications and by Helene Steffen in the protein preparations is gratefully acknowledged. This work was financially supported by the Deutsche Forschungsgemeinschaft (DFG, GA377/13) and by the Volkswagen-Foundation (I/82 627).

