

Fenoprofen and Ketoprofen Amides as Potential Antitumor Agents
Current address: Lana Pejnović, University Hospital for Infectious Diseases, Mirogojska c. 8, HR-10000 Zagreb, Croatia
Abstract
Following numerous experimental observations that various non-steroidal anti-inflammatory drugs have antitumor potentials, a series of fenoprofenamides (1a–g) and ketoprofenamides (2a–c) was tested on proliferation of different human tumor cell lines and normal human fibroblasts in vitro. Fenoprofen and ketoprofen showed modest antiproliferative activity, whereas the growth inhibitory activity of the tested amides clearly demonstrates that the substituents linked by an amide bond are essential for the significantly stronger cytostatic activity, probably because of a greater lipophilicity and/or better cell uptake. Additionally, it was shown that the most active derivatives (1d and 2a) induced cell cycle arrest at the G1 phase, as well as apoptosis.
Numerous experimental, epidemiological and clinical studies suggest that non-steroidal anti-inflammatory drugs (NSAIDs) are promising anticancer drugs (1) and may be associated with reduced risk of colon, lung, liver and other types of cancers (2). Although the mechanism responsible for the antitumor activity of NSAIDs is still unknown, it is commonly attributed to the inhibition of the inducible cyclooxygenase isoenzyme COX-2, which is overexpressed in many epithelial tumors (3). However, antineoplastic effects of NSAIDs may also include activation of apoptosis, inhibition of angiogenesis, or direct inhibition of cancer cell growth by blocking signal transduction pathways responsible for cell proliferation (4,5). Moreover, both non-selective COX-1/2 inhibitors (e.g. aspirin, sulindac, piroxicam, ibuprofen and indomethacin), as well as COX-2 selective ones (e.g. celecoxib and NS 398) have been shown to exert substantial antiproliferative effects, mainly inducing G1 cell cycle arrest or apoptosis, in various tumor cell lines regardless of COX-2 expression (6,7). Taken together, these findings suggest that NSAIDs may mediate their growth-inhibitory effects at least in part through COX-independent mechanisms.
Fenoprofen (Fen) and ketoprofen (Ket) are well-known analgesic and NSAIDs which are used in the management of mild to moderate pain, fever and inflammation processes, whereas their antitumor potential has acquired limited attention to date (8,9). Both drugs, especially Ket, have rather short plasma half-lives, therefore, repeated doses must be given to maintain the therapeutic effect (10). To minimize side-effects, prolong plasma half-life and increase water solubility or lipophilicity numerous derivates of various NSAIDs have been synthesized, which serve as potential prodrugs. For example, a number of NSAIDs derivatives such as aliphatic and aromatic esters and amides, along with amide derivatives with covalently linked anti-oxidant moieties (11–13) were prepared as potential prodrugs. Furthermore, it has been shown that a series of phenolic ester and amide derivatives of the NSAID naproxen had both antioxidative and antiproliferative activity. Besides, they were all more potent inhibitors of cell proliferation than naproxen itself and the amide derivatives tended to be more potent as antiproliferative agents than the corresponding esters (14).
In our previous papers, synthesis of Fen and Ket prodrugs of amide type was described (11,12). The present study reports the effect of fenoprofenamides and ketoprofenamides on proliferation of different human tumor cell lines, as well as normal human fibroblasts in vitro, compared with the activity of the parent compounds.
Results and Discussion
Chemistry
A series of fenoprofenamides 1a–g and ketoprofenamides 2a–c, investigated in present study, (Figure 1) were synthesized by aminolysis of Fen or Ket benzotriazolides with corresponding amine, hydroxylamine or amino acid (11,12), whereas the starting benzotriazolides were prepared from 1-benzotriazole carboxylic acid chloride and Fen or Ket, respectively (15,16).

Structural formula of fenoprofenamides 1a–g and ketoprofenamides 2a–c.
Biological results and discussion
The tested compounds showed different antiproliferative effect on the presented panel cell lines (Table 1, Figure 2). Fen and Ket showed low growth inhibitory activity at the tested concentration range, which is in agreement with the tumor cell growth-inhibitory effective concentrations of other NSAIDs in various tumor cell types published so far; the 50% inhibitory concentrations reported usually vary between 0.1 and 5 mm, with some exceptions (e.g. celecoxib) (4,17,18). However, it is clearly demonstrated that all amide derivatives of both Fen and Ket show stronger antiproliferative effect. The compounds bearing hydrophilic hydroxyl or carboxylic substituents (1f and 2c, respectively) showed little or no growth inhibition, whereas compounds 1g and 2b, both with hydroxypropyl group showed similar, slightly stronger, but still weak inhibitory activity. On the contrary, compounds 1b–e and 2a strongly and/or differently and dose-dependently inhibited the growth of all tested cell lines. Moreover, the most active ones were cyclohexyl-bearing compounds 1d and 2a (Figure 2). Comparison of all IC50 values for tumor cells and normal fibroblasts (WI38) indicates that compounds 1a, 2a and 2b showed the best selectivity – they inhibited more strongly the growth of tumor cells than the growth of normal fibroblasts. As Fen and Ket did not show any marked inhibition of cell growth at the tested concentration range, it could be concluded that the substituents are crucial for more pronounced antiproliferative activity of their amide derivatives. Given that there is obvious correlation between the biological activity and calculated lipophilicity (Clog P), one could assume that the membrane affinity/permeability may represent an important requirement for their activity. This is in correlation with other studies that showed strong correlation between the lipophilicity of various NSAIDs and their biological activity (19). Moreover, Barbato et al. showed that the lipophilicity of NSAIDs is an important prerequisite for the specific binding with COX-2, and not with COX-1 (20), which is recognized as one of the potential mechanisms of their antitumor activity.
| Compounds | IC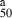 (μm) (μm) |
|||||
|---|---|---|---|---|---|---|
| Hep-2 | HeLa | MiaPaCa-2 | SW620 | MCF-7 | WI 38 | |
| Fenoprofen | >100 | >100 | >100 | >100 | >100 | >100 |
| 1a | 43 ± 2 | 44 ± 9 | ≥ 100 | 78 ± 21 | 38 ± 18 | >100 |
| 1b | 41 ± 10 | 58 ± 9 | 40 ± 0.7 | 44 ± 2 | 22 ± 7 | 48 ± 50 |
| 1c | 21 ± 0.3 | 15 ± 0.7 | 19 ± 5 | 30 ± 5 | 21 ± 1 | 16 ± 0.3 |
| 1d | 16 ± 0.1 | 19 ± 2 | 18 ± 3 | 16 ± 8 | 13 ± 8 | 21 ± 6 |
| 1e | 35 ± 14 | 18 ± 3 | 17 ± 3 | 25 ± 7 | 27 ± 2 | 27 ± 6 |
| 1f | >100 | ≥100 | >100 | >100 | >100 | >100 |
| 1g | 95 ± 50 | 58 ± 46 | >100 | 70 ± 16 | ≥100 | 36 ± 15 |
| Ketoprofen | >100 | >100 | >100 | >100 | >100 | >100 |
| 2a | 15 ± 1.7 | 17 ± 6 | 16 ± 2 | 20 ± 0.2 | 13 ± 1 | 34 ± 28 |
| 2b | 6 ± 18 | 69 ± 26 | >100 | >100 | 81 ± 20 | >100 |
| 2c | >100 | 54 ± 42 | >100 | >100 | >100 | 22 ± 6 |
- aIC50, the concentration that causes a 50% reduction of the cell growth.

Dose-response profiles for fenoprofen, ketoprofen, 1d and 2a tested on various human tumor cell lines and normal fibroblasts in vitro. The cells were treated with the compounds at different concentrations, and percentage of growth (PG) was calculated. Each point represents a mean value of four parallel samples in three individual experiments.
As compounds 1d and 2a showed the most outstanding activity, we tested them additionally, along with Fen and Ket, to check whether these compounds could induce any cell cycle perturbations and/or apoptosis in colon (SW620) and laryngeal (Hep-2) tumor cell lines. Namely, various studies thus far have reported that NSAIDs inhibit growth of human tumor cells mainly via G0/G1 cell-cycle arrest (18,21) and can also induce apoptotic cell death after a prolonged period of incubation and/or by treatment with higher concentrations (22). The treatment with Fen and Ket did not induce any difference in the distribution of cell cycle phases; neither there was an increase of the percentage of dead cells after the treatment with above mentioned compounds at c = 50 μm (data not shown). On the contrary, the treatment with compounds 1d and 2a at the same concentration, which is slightly above the IC50 concentration, had reasonable effect on the cell cycle, demonstrating the cell growth arrest in G0/G1 phase and reduction of cells in S phase of the cell cycle (Table 2). Although this effect is not spectacular, it should be stressed out that the concentration is quite low comparing to published data on cell cycle changes induced by various NSAIDs. For example, Shiff et al. have shown that the treatment with 400–1 500-μm aspirin, piroxicam, naproxen and indomethacin caused a concentration-dependant increase in the percentage of cells in G0/G1 phase and a decrease in the proportion of cells in S phase, which were noted as early as 48 h after the treatment (18). This study is entirely in accordance with ours, except that we used much lower concentration range. Higher concentrations would certainly induce more prominent effect, but as mentioned in the Introduction, it is our main goal to prepare potential antitumor compounds which should be used at as low as possible concentrations and thus induce minimal side-effects. The effects on SW620 tumor cells were visible already 24 h after the treatment with both compounds, being most drastic after 48 h, somewhat stronger for 2a than 1d. After 72 h of incubation with 1d this effect diminished, probably because a certain number of cells survived and continue to divide, whereas compound was either exhausted or metabolized by the cells during this time period. Similar observation was reported by Shiff et al., who found that the effect of piroxicam and naproxen dissipated after the first 48 h of incubation with these compounds, which could be because of the emergence of a resistant subpopulation of cells (18). The treatment of Hep-2 cells exhibited no cell cycle changes 24 h after incubation with both compounds, but cells were arrested in G1 phase during the next 48 h. Besides, 2a displayed the strongest effect (G1 arrest and S phase reduction) after 3 days of incubation. Moreover, the treatment of Hep-2 tumor cells with 1d and 2a yielded a similar and prominent increase in the percentage of dead/apoptotic cells (the subG1 population) during the 72-h period (about two times more dead cells comparing to the non-treated cells), whereas the subG1 population of SW620 cells did not differ significantly from control samples. This fact obligated us to verify the potential activation of apoptosis in Hep-2 cells by more specific test. Indeed, the Annexin V-assay confirmed that about two to four times more cells entered apoptosis after 48 and 72 h, when compared with control samples (Table 3). However, these results suggest that the G1 arrest is the major growth-inhibitory mechanism of these NSAID amides at the 50 μm concentration and that apoptosis is activated to a lesser extent and after a prolonged period of treatment. These results are in a clear accordance with the previously reported potentials of various NSAIDs to inhibit tumor cell proliferation by inducing the G1 arrest in the cell cycle and/or apoptosis (6,7,18).
| Treatmenta | Cell cycle phaseb | Hep-2 | SW620 | ||||
|---|---|---|---|---|---|---|---|
| 24 h | 48 h | 72 h | 24 h | 48 h | 72 h | ||
| Control | SubG1 | 10 | 8 | 13 | 4 | 5 | 5 |
| G0/G1 | 45 | 52 | 54 | 49 | 42 | 48 | |
| S | 23 | 28 | 31 | 37 | 46 | 37 | |
| G2/M | 32 | 20 | 15 | 13 | 12 | 15 | |
| 1d | SubG1 | 14 | 15* | 29* | 6 | 5 | 6 |
| G0/G1 | 46 | 55* | 59* | 55* | 52* | 54* | |
| S | 24 | 28 | 26* | 30* | 38* | 32* | |
| G2/M | 30 | 17 | 15 | 15 | 10 | 14 | |
| 2a | SubG1 | 11 | 15* | 30* | 5 | 5 | 4 |
| G0/G1 | 46 | 54* | 66* | 53* | 57* | 48 | |
| S | 28 | 26 | 19* | 32* | 32* | 40 | |
| G2/M | 26 | 20 | 15 | 15 | 11 | 12 | |
- a c = 50 μm.
- bThe results are shown as percentages of cell population in each cell cycle phase. The experiment was repeated three times, and the results were within 10%.
- *Statistically significant at p < 0.05.
| Treatmenta | Time (h) | ||
|---|---|---|---|
| 24 | 48 | 72 | |
| Control | 1.9 ± 1.8 | 0.9 ± 0.4 | 2.0 ± 1.7 |
| 1d | 2.1 ± 0.5 | 3.5 ± 0.6 | 4.7 ± 0.5 |
| 2a | 0.7 ± 0.1 | 5.3 ± 2.2 | 5.8 ± 0.5 |
- a c = 50 μm.
Conclusions and Future Directions
Following numerous experimental observations that various NSAIDs have antitumor potentials, a series of Fen and Ket amide derivatives was tested on proliferation of different human tumor cell lines and normal human fibroblasts in vitro. Fen and Ket showed modest antiproliferative activity, whereas the growth inhibitory activity of the tested amides clearly demonstrates that the substituents linked by an amide bond are essential for the significantly stronger cytostatic activity, probably because of a greater lipophilicity and/or better cell uptake. Additionally, it was shown that the most active derivatives (1d and 2a) induced cell cycle arrest at the G1 phase, as well as apoptosis, which are major mechanisms of NSAIDs antitumor activity. We believe that these investigations should form the basis for further research and synthetic optimization of novel NSAID amides as potential prodrugs for antitumor therapy or chemopreventive applications with less-toxic side effects. Currently, studies are in progress to assess the anti-inflammatory activity of these compounds, as well as COX selectivity.
Experimental Section
Chemistry
Fenoprofenamides 1a–g and ketoprofenamides 2a–c were synthesized following previously published procedures (11,12). The structural formula of the prepared amides is given in Figure 2. All analytical and spectral data were in agreement with the previously published results. Fen was purchased from Eli Lilly Company, (Indianapolis, IN, USA), Ket was kindly obtained from Belupo (Croatia) and all amines were purchased from Aldrich (St.Louis, MO, USA). The octanol-water partition coefficients (log P) were calculated by ChemDraw Ultra 6.0 (CambridgeSoft Corporation, Cambridge, MA, USA).
Biological studies
Proliferation assay
The HeLa, MiaPaCa-2, SW620, MCF-7, Hep-2 and WI 38 cell lines were seeded into a series of standard 96-well microtiter plates on day 0, at 1 × 104 to 3 × 104 cells/mL, depending on the doubling times of the specific cell lines. Test compounds were then added in five, 10-fold dilutions (10−8–10−4 m) and incubated for a further 72 h. Stock solutions were prepared in DMSO, (c = 0.1 m), whereas working dilutions were freshly prepared on the day of testing. The solvent (DMSO) was also tested for eventual inhibitory activity by adjusting its concentration to be the same as in working concentrations (DMSO concentration never exceeded 0.1%). After 72 h of incubation, the cell growth rate was evaluated by performing the MTT assay, as described previously (9).
Cell cycle analysis
Cells were seeded (2 × 105 per well) in a 6-well plate. After 24 h the tested compounds were added at concentration of 50 μm. The attached cells were trypsinized, combined with floating cells, washed with phosphate buffer saline (PBS) and fixed in 70% ethanol for 24, 48 and 72 h after the treatment with compounds. Immediately before the analysis, the cells were washed with PBS and stained with 2.5 μg/mL of propidium iodide (PI) with the addition of 0.2 μg/μL of RNAse A. The stained cells were then analyzed with FACSCalibur™ (Becton Dickinson, Immunocytochemistry Systems, San Jose, CA, USA) flow cytometer (20 000 counts were measured). The percentage of the cells in each cell cycle phase was determined using modfit ltTM software (Verity Software House Inc, Topsham, ME, USA) based on the DNA histograms. As a minimum, three experiments were carried out in triplicates, and Student's t-test (p < 0.05) was used to measure the statistical significance.
Annexin-V test
Detection and quantification of apoptotic cells at single cell level were performed using Annexin-V-FLUOS staining kit (Roche Diagnostic GmbH, Mannheim, Germany), according to the manufacturer's recommendations. After a desired length of time, both floating and attached cells were collected. The cells were then washed with PBS, pelleted and resuspended in staining-solution [annexin-V-fluorescein labeling reagent and PI in Hepes buffer]. The cells were then analyzed under a fluorescence microscope. Annexin-V (green fluorescent) cells were determined to be apoptotic. Percentage of apoptotic cells was expressed as a number of fluorescent cells in relation to the total cell number (fluorescent and non-fluorescent cells), which was expressed as 100%.
Acknowledgments
The financial support of the Ministry of Science, Education and Sports of the Republic of Croatia is gratefully acknowledged.

