

Maternally administered melatonin protects against ischemia and reperfusion-induced oxidative mitochondrial damage in premature fetal rat brain
Abstract
Abstract: We investigated the oxidative susceptibility of the brain and the effect of maternally administered melatonin on ischemia/reperfusion-induced cerebral damage in premature fetal rat. Fetal brain mitochondria was separated on the 16th and 19th days of pregnant rats and the respiratory control index (RCI) was measured as an indicator of mitochondrial respiratory activity in the presence or absence of xanthine and xanthine oxidase. The utero–ovarian arteries were occluded bilaterally for 20 min in female rats on day 16 of pregnancy to induce fetal ischemia. Reperfusion was achieved by releasing the occlusion and restoring circulation for 30 min. A sham operation was performed in control rats. Melatonin (10 mg/kg) or vehicle was injected intraperitoneally into the dams 60 min prior to occlusion. The RCI and concentration of thiobarbituric acid-reactive substances (TBARS) in fetal brain mitochondria were measured. The addition of xanthine and xanthine oxidase significantly decreased mitochondrial RCI at both the 16- and 19-day-old fetal brain. Xanthine and xanthine oxidase-induced reduction in RCI was significantly greater in the 16-day-old fetal brain than that in the fetal brain from the 19th day of pregnancy. Ischemia/reperfusion significantly reduced RCI and elevated TBARS concentrations in the 16-day-old fetal brain mitochondria. Melatonin treatment reversed ischemia/reperfusion-induced reduction in RCI (2.22 ± 0.10 to 2.53 ± 0.08, P < 0.01) and elevation in TBARS concentrations (13.50 ± 1.82 nmol/mg protein to 8.80 ± 0.78 nmol/mg protein, P < 0.01), resulting in values similar to those in untreated, sham-treated animals. Results indicate that brain mitochondria in the premature fetal rats appear to be more susceptible to oxidative damage. Melatonin administration to pregnant rats may prevent ischemia/reperfusion-induced oxidative mitochondrial damage in premature fetal brain.
Introduction
Fetal neurological deterioration is believed to occur as a result of asphyxia during prolonged labor [1]. However, intrapartum asphyxia accounts for less than 10% of cases of cerebral damage [2, 3], and most neurological insults occur prenatally [4, 5]. Oxygen free radicals produced during fetal development can induce oxidative damage of brain tissue. We previously demonstrated that ischemia and subsequent reperfusion of the utero–ovarian arteries stimulates the activity of xanthine oxidase or lipoxygenase [6, 7], which may cause oxidation of cerebral lipid and DNA in the fetus [8]. In addition, ischemia/reperfusion induces oxidative damage of mitochondria in the fetal brain, which may ultimately lead to the impairment of fetal central nervous system [9].
Melatonin, secreted by the pineal gland, is a powerful scavenger of oxygen free radicals, quenching hydroxyl radicals (OH·) [10–14] and possibly, peroxyl radicals (LOO·) as well [15]. We previously reported that melatonin is rapidly transferred from maternal to fetal circulation and brain tissue in rats [16, 17]. Maternally administered melatonin prevents oxidative lipid, DNA, and mitochondrial damage in mature fetal rat brain [8, 18]. In addition, the administration of melatonin to pregnant rats increases the activities of superoxide dismutase (SOD) and of glutathione peroxidase (GSH-Px) in near-term fetal rat brain [17]. In clinical practice, however, the incidence of cerebral damage is more frequent in premature fetus. Brain tissue is highly sensitive to damage by oxygen free radicals because of its high utilization of oxygen, its high concentration of polyunsaturated fatty acids (PUFAs) and of transition metals such as iron, and its low concentration of cytosolic antioxidants [19]. Inder et al. [20] have shown elevated levels of free-radical products in the cerebrospinal fluid of premature infants with cerebral white matter injury. It is therefore likely that premature fetal brain is more susceptible to oxidative damage by oxygen free radicals.
In the present study, we evaluated the oxidative susceptibility of the brain and investigated whether administration of melatonin to the mother would prevent ischemia/reperfusion-induced oxidative cerebral damage in premature fetal rat.
Materials and methods
Wistar rats were maintained in accordance with the Kochi Medical School Guidelines for the Care and Use of Laboratory Animals. Animals were housed in plastic cages at a constant temperature of 24 ± 2°C. The light : dark cycle was maintained at 14 : 10 hr (lights on, 05:00 hr, lights off, 19:00 hr).
Experimental protocol 1
Fetal brain tissues were obtained from day 16 and 19 pregnant rats and homogenized in medium consisting of 0.24 m mannitol, 0.06 m sucrose, 50 μm ethylenediaminetetraacetic acid (EDTA), and 5 mm Tris–HCl, pH 7.2. After the fetal brain homogenate was centrifuged at 490 × g for 5 min, the supernatant was collected and centrifuged at 7800 × g for 10 min. The resulting pellet was suspended in a medium consisting of 0.25 m sucrose and 1 mm Tris–HCl (pH 7.2), and used as the brain mitochondrial fraction [7]. All procedures were carried out at 0–4°C. Mitochondrial respiratory control index (RCI) was measured with or without 50 μmol/mL xanthine and 0.02 units/mL xanthine oxidase. Each group consists of seven pregnant rats.
Experimental protocol 2
Laparotomy was performed on pregnant rats on day 16 of pregnancy after injection of ketamine (40 mg/kg, i.p.) and xylazine (10 mg/kg, i.p.). Fetal ischemia was induced by occluding the utero–ovarian artery bilaterally for 20 min, using forceps covered with soft polyvinyl tubing. Reperfusion was achieved by releasing the clamp from utero-ovarian arteries to restore circulation for 30 min (n = 12). Complete interruption and restoration of blood flow were confirmed directly using an operating microscope. Control rats underwent a sham operation that omitted vascular clamping (n = 15). Melatonin was dissolved in a minimal volume of absolute ethanol and diluted to 2.5 mg/mL with physiological saline. The concentration of ethanol in the final solution was 5%. We previously reported that administration of melatonin to pregnant rats markedly increased the levels of melatonin in maternal plasma and fetal brain, with the maximal level attained at 60 min after administration [17]. Therefore, we injected the melatonin solution i.p. at 10 mg/kg, 60 min prior to occlusion of the utero–ovarian arteries (n = 8). Rats not treated with melatonin were injected with an equal volume of vehicle. All experimental procedures were performed between 15:00 and 18:00 hr. At the end of the experimental period, RCI and the concentrations of thiobarbituric acid-reactive substances (TBARS) were measured in fetal brain mitochondria.
Determination of mitochondrial respiratory activity
Oxygen consumption was measured polarographically at 25°C using 1.0–2.5 mg of protein from the fresh mitochondrial fraction in 2.0 mL of incubation medium consisting of 100 mm KCl, 0.05 mm EDTA, 10 mm Tris–HCl, and 0.1 m sucrose at pH 7.4, using a Clark-type electrode. Mitochondrial respiration was initiated by adding 150 μm adenosine 5-diphosphate (ADP) with 5 mm succinate as the respiratory substrate. Oxygen consumption measured in the presence of added ADP was defined as state 3 respiration, while that measured following consumption of ADP was defined as state 4 respiration. The RCI, calculated as a ratio of state 3 to state 4 respiration, was used as a marker of mitochondrial respiratory activity.
Determination of TBARS concentrations
The concentration of TBARS in fetal brain mitochondria was determined according to the method of Ohkawa et al. [21]. Aliquots containing 1.5 mg of protein were mixed with 100 μL 8.1% of sodium dodecyl sulfate (SDS). Then, 1.5 mL of 20% acetic acid (pH 3.5) and 1.5 mL of a 0.8% TBA solution were added to the mixture, and the volume was adjusted to 4.0 mL by addition of distilled water. The mixture was shaken thoroughly and then heated in an oil bath at 95°C for 60 min. After cooling the mixture with tap water, 1.0 mL of distilled water and 5.0 mL of butyl alcohol and pyridine (15 : 1, v/v) were added. The mixture was shaken gently for 5 min. After centrifugation at 1500 × g for 10 min, the butyl alcohol–pyridine phase containing the TBARS was separated and its absorbance was measured at 532 nm. The results are expressed as the molar equivalent of malondialdehyde per mg of protein, using malondialdehyde from tetramethoxypropane as the standard and double-distilled water as the control.
Statistical analysis
All data are expressed as the mean ± S.E. Xanthine and xanthine oxidase-induced changes in RCI were analysed by Student's paired t-test. Differences in xanthine oxidase-induced changes in RCI between 16- and 19-day-old fetal rat brain were determined by Student's unpaired t-test. One-way analysis of variance (ANOVA) was used to compare RCI and concentration of TBARS among the three groups of animals: untreated sham–ischemic animals; untreated animals subjected to ischemia for 20 min followed by reperfusion for 30 min; and animals treated with melatonin prior to ischemia/reperfusion. If the ANOVA showed a significant difference, Scheffe's multiple comparison procedure was applied to determine which values differed. A level of P < 0.05 was accepted as statistically significant.
Results
The mitochondrial RCI was significantly reduced by the addition of xanthine plus xanthine oxidase in both 16- and 19-day-old fetal brains. A decrease in RCI by the addition of xanthine and xanthine oxidase in the 16-day-old fetal brain was significantly greater than that in the 19-day-old fetal brain (Fig. 1).

(A) Mitochondrial respiratory control index in the 16- and 19-day-old fetal rat brains. Closed and open columns represent with and without xanthine plus xanthine oxidase, respectively. (B) Xanthine and xanthine oxidase-induced reduction of mitochondrial respiratory control index in the 16- and 19-day-old fetal rat brains.
Significant differences among the three groups existed for mitochondrial RCI and TBARS concentrations in the fetal brain. Ischemia and subsequent reperfusion significantly reduced mitochondrial RCI, while significantly increasing TBARS concentrations (Fig. 2). Maternally administered melatonin, however, reversed the ischemia/reperfusion-induced decrease in RCI from 2.22 ± 0.10 to 2.53 ± 0.08 and increase in TBARS concentrations from 13.5 ± 1.82 to 8.80 ± 0.78 nmol/mg protein (Fig. 2). Mitochondrial RCI and TBARS concentrations in the melatonin group did not differ significantly from the sham-operated control group.

(A) Mitochondrial respiratory control index in the 16-day-old fetal rat brain. Open, closed, and diagonally hatched columns represent sham-operated control, ischemia–reperfusion, and melatonin-treated groups, respectively. NS, not significant. (B) Thiobarbituric acid-reactive substances concentrations in the 16-day-old fetal rat brain mitochondria. Open, closed, and diagonally hatched columns represent sham-operated control, ischemia–reperfusion, and melatonin-treated groups, respectively.
Discussion
In the present study, to evaluate the oxidative susceptibility of fetal brain mitochondria induced by oxygen free radicals, we measured mitochondrial respiratory activities in the presence or absence of xanthine and xanthine oxidase. Xanthine is metabolized to uric acid by xanthine oxidase. During the degradation of xanthine, oxygen free radicals, especially O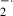 were produced. The mitochondrial RCI, an index of mitochondrial respiratory activity, was decreased by the addition of xanthine and xanthine oxidase in the 16- and 19-day-old fetal brains. O
were produced. The mitochondrial RCI, an index of mitochondrial respiratory activity, was decreased by the addition of xanthine and xanthine oxidase in the 16- and 19-day-old fetal brains. O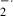 has been reported to increase free iron by oxidizing iron–sulfur clusters, such as aconitase in the mitochondria [22]. Accordingly, the O
has been reported to increase free iron by oxidizing iron–sulfur clusters, such as aconitase in the mitochondria [22]. Accordingly, the O generated by the addition of xanthine and xanthine oxidase may be rapidly converted to highly toxic OH· and may impair mitochondrial function of fetal brain in both mature and premature fetal rats. However, oxygen free-radical-induced decrease in mitochondrial RCI was significantly greater in the 16-day-old fetal brain than in the 19-day-old fetal brain, indicating that premature fetal brain is more susceptible to oxidative damage. A clinical study has also shown increased lipid and protein oxidation products in the cerebrospinal fluid of premature human infants with cerebral white matter injury [20]. Although the mechanisms for the high oxidative susceptibility of premature fetal brain are unclear, Houdou et al. [23] have demonstrated a reduced catalase expression in white matter glial cells until approximately 31 wk of gestation. This evidence suggests that the defense against the damage induced by oxygen free radicals may be underdeveloped in immature fetal brain.
generated by the addition of xanthine and xanthine oxidase may be rapidly converted to highly toxic OH· and may impair mitochondrial function of fetal brain in both mature and premature fetal rats. However, oxygen free-radical-induced decrease in mitochondrial RCI was significantly greater in the 16-day-old fetal brain than in the 19-day-old fetal brain, indicating that premature fetal brain is more susceptible to oxidative damage. A clinical study has also shown increased lipid and protein oxidation products in the cerebrospinal fluid of premature human infants with cerebral white matter injury [20]. Although the mechanisms for the high oxidative susceptibility of premature fetal brain are unclear, Houdou et al. [23] have demonstrated a reduced catalase expression in white matter glial cells until approximately 31 wk of gestation. This evidence suggests that the defense against the damage induced by oxygen free radicals may be underdeveloped in immature fetal brain.
Ischemia and subsequent reperfusion of utero-ovarian arteries increased TBARS concentrations, while they reduced mitochondrial RCI in the 16-day-old fetal brain, indicating that oxygen free radicals produced by ischemia/reperfusion may induce mitochondrial lipid peroxidation in premature fetal brain. We have previously clarified several mechanisms of fetal cerebral oxidative damage induced by ischemia and reperfusion. Ischemia/reperfusion stimulates the activity of lipoxygenase in fetal rat brain, which may lead to peroxidation of arachidonic acid [7]. Ischemic insults cause the accumulation of hypoxanthine and stimulate xanthine oxidase activity, while subsequent reperfusion enhances degradation of hypoxanthine to uric acid in fetal rat brain [6]. As xanthine oxidase is a major potential source of oxygen free radicals, especially O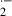 , ischemia/reperfusion may stimulate the generation of xanthine oxidase-derived O
, ischemia/reperfusion may stimulate the generation of xanthine oxidase-derived O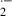 , which can result in lipid peroxidation of mitochondrial membranes. In contrast, maternally administered melatonin to pregnant rats prevented increases in concentration of TBARS in fetal brain mitochondria resulting from ischemia/reperfusion [8]. In addition, melatonin reversed ischemia/reperfusion-induced reduction in mitochondrial respiratory activity [24]. These results indicate that maternally administered melatonin can cross the placenta [16] to reach the fetal brain [17] where it quenches ischemia/reperfusion-induced oxygen free radicals to prevent cerebral mitochondrial lipid peroxidation in premature fetal rat.
, which can result in lipid peroxidation of mitochondrial membranes. In contrast, maternally administered melatonin to pregnant rats prevented increases in concentration of TBARS in fetal brain mitochondria resulting from ischemia/reperfusion [8]. In addition, melatonin reversed ischemia/reperfusion-induced reduction in mitochondrial respiratory activity [24]. These results indicate that maternally administered melatonin can cross the placenta [16] to reach the fetal brain [17] where it quenches ischemia/reperfusion-induced oxygen free radicals to prevent cerebral mitochondrial lipid peroxidation in premature fetal rat.
Melatonin is reported to be a strong oxygen free radical scavenger of OH·, which initiates lipid peroxidation [10–14]. Melatonin may also scavenge LOO·, which propagates lipid peroxidation [15]. According to Cuzzocrea et al. [25] and Gilad et al. [26], melatonin scavenges the high toxic peroxynitrite anion, which is a reaction product of superoxide and nitric oxide and may induce mitochondrial oxidative damage. In addition, melatonin has been reported to inhibit the activity of cerebral nitric oxide synthase [27, 28], and also scavenge nitric oxide directly [29, 30], thereby decreasing the production of peroxynitrite anion. We previously demonstrated that melatonin administration to the mother increases the activity of SOD as well as GSH-Px in fetal rat brain [17, 18]. In addition, melatonin also increases levels of mRNA for GSH-Px, copper–zinc SOD, and manganese SOD in rat cerebral cortex [31, 32]. Although we did not measure the activities of antioxidant enzymes such as SOD or GSH-Px in the present study, it is possible to speculate that melatonin may protect against oxidative cerebral damage by increasing these activities in addition to its direct antioxidant effect.
Cerebral white matter injury such as periventricular leukomalacia (PVL) contributes significantly to neonatal mortality and long term neurodevelopmental deficits in premature infants. PVL has been reported to be associated with an increased risk of cerebral palsy [33]. Although the pathogenesis of white matter injury is complex and multifactorial, oxygen free radical-induced oxidative damage appears to be important [34]. Clinical studies are required to determine whether melatonin can protect premature fetal brain against oxidative impairment occurring during pre-eclampsia and other disorders characterized by increased oxidative stress.

