

Hermansky–Pudlak syndrome: a disease of protein trafficking and organelle function
Summary
The Hermansky–Pudlak syndrome (HPS) is a collection of related autosomal recessive disorders which are genetically heterogeneous. There are eight human HPS subtypes, characterized by oculocutaneous albinism and platelet storage disease; prolonged bleeding, congenital neutropenia, pulmonary fibrosis, and granulomatous colitis can also occur. HPS is caused primarily by defects in intracellular protein trafficking that result in the dysfunction of intracellular organelles known as lysosome-related organelles. HPS gene products are all ubiquitously expressed and all associate in various multi-protein complexes, yet HPS has cell type-specific disease expression. Impairment of specialized secretory cells such as melanocytes, platelets, lung alveolar type II epithelial cells and cytotoxic T cells are observed in HPS. This review summarizes recent molecular, biochemical and cell biological analyses together with clinical studies that have led to the correlation of molecular pathology with clinical manifestations and led to insights into such diverse disease processes such as albinism, fibrosis, hemorrhage, and congenital neutropenia.
Introduction
In 1959, the Czechoslovakian physicians Hermansky and Pudlak described two patients with oculocutaneous albinism, prolonged bleeding and pigmented macrophages in the bone marrow; one patient also had interstitial pulmonary fibrosis and died at 34 yr of age (Hermansky and Pudlak, 1959). Since that time, the Hermansky–Pudlak syndrome (HPS), has been recognized as a genetically heterogeneous set of related autosomal recessive conditions due to mutations in genes that mostly function in membrane and protein trafficking. Defects in proteins encoded by these genes can affect the biogenesis and/or function of intracellular organelles found in specialized secretory cells such as pigment cells (i.e. melanocytes and pigment epithelial cells), platelets, T cells, neutrophils, and lung type II epithelial cells. The organelles affected by HPS genes belong to the family of organelles known as lysosome-related organelles (LROs), which share in common with lysosomes at least one integral membrane protein and intralumenal acidic pH (Cutler, 2002; Dell'Angelica et al., 2000b). Although HPS disease phenotype appears to be confined to only certain specialized cell types, expression of HPS genes has been detected in all tissues tested, and HPS genes are thought to be ubiquitously expressed.
There are eight known human HPS genes, each of which can lead to a particular clinical HPS subtype. There are 15 murine HPS genes, which have been cloned and sequenced; eight of the mouse genes are orthologous to the eight human HPS genes (Table 1), and many HPS genes have orthologues in the Drosophila melanogaster genome as well (Dell'Angelica, 2004). It is likely that additional human HPS subtypes will be described, corresponding to the known mouse strains.
| Human subtype | Mouse strain | Gene | Function |
|---|---|---|---|
| HPS-1 | Pale ear | HPS1 | ? |
| HPS-2 | Pearl | AP3B1 | Cargo selection and vesicle trafficking to the lysosome |
| HPS-3 | Cocoa | HPS3 | ? |
| HPS-4 | Light ear | HPS4 | ? |
| HPS-5 | Ruby eye-2 | HPS5 | ? |
| HPS-6 | Ruby eye | HPS6 | ? |
| HPS-7 | Sandy | DTNBP1 | Binds α- and β-dystrobrevin, myospryn |
| HPS-8 | Reduced pigmentation | BLOC1S3 | ? |
| ? | Pallid | Pldn | Binds syntaxin 13 |
| ? | Muted | mu | ? |
| ? | Cappuccino | cno | ? |
| ? | Buff | VPS33A | Vesicle trafficking to yeast vacuole |
| ? | Gunmetal | RABGGTA | Adds lipophilic prenyl groups to carboxyl terminus of rab proteins |
| ? | Mocha | AP3D1 | Cargo selection & vesicle trafficking to the lysosome |
| ? | Subtle gray | Slc7a11 | Cystine/glutamate exchanger xCT |
- AP3B1, adaptor protein complex-3 β3A subunit; DTNBP1, dystrophin binding protein-1, also known as dysbindin; Pldn, pallidin; RABGGTA, Rab geranylgeranyl transferase α subunit; AP3D1, adaptor protein complex-3 δ-subunit, ? denotes unknown.
Of the 15 identified HPS genes, only five have known functions (AP3B1, AP3D1, VPS33A, RABGGTA, and Slc7a11), and four of these have roles in regulating membrane/vesicle and protein trafficking. The AP3B1 gene encodes the β subunit and AP3D1 encodes the δ subunit of the adaptor protein AP-3, which plays a role in enriching cargo proteins in vesicles for transport through the intracellular endosomal/lysosomal pathway. The VPS33A gene encodes a protein that is part of the class C Vps complex (C-Vps complex) which plays an integral role in vesicular trafficking to the yeast vacuole, an organelle functionally homologous to the mammalian lysosome (Suzuki et al., 2003), by binding a syntaxin homolog to mediate transport vesicle docking and fusion with a target membrane such as the vacuolar membrane (Sato et al., 2000; Suzuki et al., 2003). The C-Vps complex is also a component of the larger HOPS complex which is essential for vacuole to vacuole fusion and vacuole protein sorting (Seals et al., 2000). The RABGGTA gene product Rab geranylgeranyl transferase-α is a subunit of a heterodimer that attaches prenyl moieties to Rab molecules, which are small GTP-binding proteins in the Ras-like GTPase superfamily which regulate vesicular trafficking and organelle motility (Detter et al., 2000).
Two other HPS gene products are known to bind to previously described proteins. The dystrobrevin binding protein-1 (DTNBP1) gene product is dysbindin, that binds to α- and β-dystrobrevins, components of the dystrophin-associated protein complex (Benson et al., 2001), which is found at the synapses in brain and muscle cells (Sillitoe et al., 2003); dysbindin has been suggested to play a role in the exocytosis of glutamate in neuronal cells (Numakawa et al., 2004). The Pldn gene product, pallidin, binds to syntaxin 13 in yeast two hybrid studies (Huang et al., 1999). Syntaxin 13 is a member of the SNARE family of molecules that mediate membrane docking and fusion (Advani et al., 1998); the interaction of syntaxin 13 with pallidin suggests a role for pallidin in membrane trafficking.
The only HPS gene of known function that does not have an identified role in protein or membrane trafficking, Slc7a11, is defective in the subtle gray mouse strain, which has been classified as a model for mild HPS because of mild depression of platelet dense granule numbers by electron microscopic examination (Swank et al., 1996). Slc7a11 encodes the cystine/gutamate exchanger xCT (Chintala et al., 2005). This protein appears to regulate the ratio of pheomelanin and melanin synthesized in melanocytes, and affects cells’ ability to respond to oxidative stress.
Strikingly, many of the HPS proteins have one or two predicted regions of coiled coil motif, which have been implicated in protein–protein interactions (Burkhard et al., 2001; Dell'Angelica, 2004). Furthermore, all of the HPS proteins are associated in multi-protein complexes, the majority involving multiple HPS proteins (Table 2). Thus, biogenesis of lysosome-related organelle complex-1 (BLOC-1) includes as subunits the HPS proteins pallidin, cappuccino, muted, reduced pigmentation (rp) and dysbindin (Ciciotte et al., 2003; Falcon-Perez et al., 2002; Gwynn et al., 2004; Starcevic and Dell'Angelica, 2004); BLOC-2 has as subunits the HPS3, HPS5, and HPS6 proteins (Di Pietro et al., 2004; Gautam et al., 2004; Zhang et al., 2003); BLOC-3 contains HPS1 and HPS4 proteins (Chiang et al., 2003; Martina et al., 2003; Nazarian et al., 2003). The adapter protein complex AP-3 includes the β3A subunit, defective in HPS-2 (Dell'Angelica et al., 1999) and the δ-subunit, defective in the murine HPS mocha strain (Kantheti et al., 1998). Two other proteins, VPS33A and RGGTA, are defective in murine HPS, and are also known to form multiprotein complexes (Detter et al., 2000; Rieder and Emr, 1997; Seals et al., 2000) but no human diseases have yet been found to be caused by defects in these proteins.
| Complex | Subunits |
|---|---|
| AP-3 | β3A δμ3σ3 |
| BLOC-1 | Pallidin Cappuccino Muted Reduced pigmentation/BLOS3aDysbindin Snapin BLOS1 BLOS2 |
| BLOC-2 | HPS3 HPS5 HPS6 |
| BLOC-3 | HPS1 HPS4 |
| Predicted complex | Predicted subunits |
| VpsC complex | Vps33a, Vps11, Vps16, Vps18 |
| Rab geranylgeranyl transferase | Rabggta, Rabggtb |
- aMurine HPS8.
Clinically, HPS is defined by pigment dilution (affecting skin, hair, and eyes) – resulting in oculocutaneous albinism – and platelet storage pool deficiency (causing prolonged bleeding), but different HPS subtypes have additional distinguishing features, discussed in subsequent sections. Some subtypes, notably HPS-1 and HPS-4, can be debilitating and can lead to premature mortality; there is no known cure or effective therapy for HPS. A diagnosis of HPS can be made by (1) an ophthalmologic examination showing iris transillumination, fundus hypopigmentation, the presence of nystagmus and decreased visual acuity; and (2) a wet mount electron microscopic examination of platelets showing absent or greatly decreased numbers of platelet dense granules (organelles that store ATP, ADP, calcium, serotonin for release upon platelet aggregation) (Huizing and Gahl, 2002). Accumulation of an autofluorescent ceroid-like material can be detected in the reticuloendothelial system (Nakatani et al., 2000; White et al., 1973; Witkop et al., 1989). Confirmation of the diagnosis and subtyping is done by molecular analysis demonstrating mutation of an HPS gene.
Even in cases in which mutations in HPS genes are identified, ascertaining and attributing gene effects can sometimes be tentative due to low numbers of patients (in some HPS subtypes only one to three patients have been identified) and background gene effects (i.e. mutations or polymorphisms in other genes) can be difficult to control for. Studying the mouse models of HPS can be illuminating, as most of the HPS subtypes occurred spontaneously in or were bred subsequently onto a common inbred genetic background (C57BL/6), and gene effects can be more reliably identified. However, the mouse strains do not harbor the same mutations as seen in human patients, so phenotypes occurring in humans may not be represented in the mouse strains. The study of both HPS affected patients and mice has yielded complementary information that has recently allowed a better understanding of the pathogenesis and cellular basis of HPS, and has spurred interest in HPS in diverse fields such as dermatology, ophthalmology, pulmonology, hematology, genetics and cell biology.
HPS-1
Gene and protein structure
The human HPS1 gene spans 30.5 kb on chromosome segment 10q23.1–q23.3 and has 9730 base pairs encoding 20 exons, the first two of which are noncoding (Bailin et al., 1997); a major 3.0 kb and minor 3.9 kb mRNA (attributed to the use of proximal and distal polyadenylation sites) have been demonstrated (Oh et al., 1996; Wildenberg et al., 1998). Alternative splicing of exon 9 occurs, with up to 50% of mRNA noted to lack exon 9 (Gonzalez-Conejero et al., 2003; Oh et al., 1996). A pseudogene has been detected on chromosome 22q12.2–12.3 which has high sequence similarity to HPS1 exons 2–5 and 100% sequence homology to HPS1 exon 6 (Huizing et al., 2000).
The 700 amino acid polypeptide has a predicted MW of 79.3 kDa, is not N-glycosylated in melanoma cells (Oh et al., 2000), and is predicted to have a coiled-coil region (Dell'Angelica, 2004). A minor 75.9 kDa protein results from the alternative splicing of exon 9 (Oh et al., 2000).
Fifteen nonpathologic polymorphisms have been noted in HPS1, 3 of which cause amino acid changes: G283W, P491R, and R603Q (Bailin et al., 1997). Twenty-three disease causing mutations have been reported in HPS1 (Figure 1). The most common HPS1 mutation is found in Puerto Ricans (over 400 hundred patients have been identified) and is caused by a 16-bp frameshift duplication in exon 15 (Oh et al., 1996).

Mutations found in HPS-1 and pale ear. Oh (1998), Spritz (1999), Ito (2005), Hermos (2002), Gonzalez-Conejero (2003), Shotelersuk (1998), Natsuga (2005), Horikawa (2000), Oh (1996), Oetting (1999), Feng (1997), Gardner (1997).
Clinical features
HPS-1 is the most frequently presenting HPS subtype, the most common genetic disease among Puerto Ricans, occurring with a frequency of one in 1800 in northwest Puerto Rico (Witkop et al., 1990), and accounts for ∼50% of non-Puerto Rican cases (Oh et al., 1998). Mutations have been found in European, Japanese, Turkish and Pakistani cohorts.
The homozygous 16-bp duplication in the HPS1 gene is found exclusively in those of Puerto Rican heritage, and is associated with a severe phenotype: restrictive lung disease was found to occur in 68% of affected individuals, hemorrhage in 17%, and granulomatous colitis in 15% (Witkop et al., 1990). Recent advances in genetic testing have led to several studies detailing the specifics of the clinical phenotype resulting from the 16-bp duplication.
Forty-nine percent of patients have been noted to have abnormal chest radiographs and 82% have abnormal high resolution chest computed tomography (CT) scans, but all patients maintain normal oxygen saturation at rest (Brantly et al., 2000). Lung tissue showed cystic/honeycomb changes (Nakatani et al., 2000), similar to findings seen in HPS mouse lungs (McGarry et al., 1999), and patchy fibrosis. Cells were filled with phospholipids droplets; enlarged lamellar bodies (organelles that store surfactant protein and phospholipids prior to secretion) were increased in number, suggesting a defect in their secretion (Nakatani et al., 2000).
Onset and rate of progression of pulmonary disease can be followed with pulmonary function tests and high resolution CT (Avila et al., 2002; Brantly et al., 2000). The anti-fibrotic agent pirfenidone appears to slow the progression of lung disease in HPS1 patients with a residual forced vital capacity (FVC) > 50% (Gahl et al., 2002). The restrictive lung disease has an average age of symptomatic onset at 35 yr, leading to an average age of death at 37 yr, but there is a wide individual variability in pulmonary function, suggesting the influence of environmental triggers/exacerbators and/or modifying genes. Health maintenance measures, e.g. avoidance of primary and secondary cigarette smoke/lung toxins, prompt treatment of respiratory infections, administration of the influenza and pneumovax vaccines, were recommended (Brantly et al., 2000).
Prolonged bleeding is usually noted with the onset of walking, but can vary from easy bruisability to frequent (more than twice a month) or prolonged (>1 h) bleeding episodes to major bleeding episodes lasting >12 h requiring transfusions, hospitalization, or treatment such as cautery (Gahl et al., 1998; Hermos et al., 2002). One study, in which patients were genotyped, concluded that platelet transfusion remains the treatment of choice for HPS-1 patients (Cordova et al., 2004). All patients had absence of platelet dense granules by electron microscopic examinations (Gahl et al., 1998). Avoidance of aspirin was recommended.
The best corrected vision in HPS-1 patients ranges from 20/250 to 20/80. Iris, choroid and retinal pigmentation, while variably decreased, does not correlate with skin pigmentation. Horizontal nystagmus is present in the primary position with a rotary component often also noted. On slit lamp examination, iris transillumination is present to variable degrees, and fundus hypopigmentation and foveal hypoplasia are noted (Gahl et al., 1998).
Inflammatory bowel disease is generally diagnosed between the ages of 11 and 25 yr, and only involved the large intestine, with most patients requiring surgical resection and colostomy placement (Gahl et al., 1998).
On skin examination, there is a broad variability in hair and skin pigmentation (Gahl et al., 1998), but interestingly, as is reported for pale ear mouse coat color (the mouse model for HPS-1), the hair and skin of individuals affected with HPS-1 darkens as patients age (Lane and Green, 1967; Toro et al., 1999). Patients have melanocytic nevi, 29% have acanthosis nigricans-like epidermal changes on the neck and axilla without signs or symptoms of endogenous or iatrogenic insulin resistance, and 70% have trichomegaly of the arms, legs, and eyelashes (Toro et al., 1999).
A study of renal function in HPS-1 patients carrying the homozygous 16-bp duplication revealed no significant dysfunction in glomerular or renal tubular functions, although creatinine clearance was decreased in one-third of the HPS-1 patients with the 16-bp duplication compared with one-tenth of the HPS-1 patients without (Gahl et al., 1998).
The clinical findings in HPS-1 patients not bearing the 16-bp duplication can range from mild to severe. For example, the homozygous mutation P324ins found in Swiss patients was not associated with intracellular ceroid accumulation, pulmonary fibrosis or inflammatory bowel disease and was generally noted to result in a mild HPS phenotype (Frenk and Lattion, 1982; Oh et al., 1996; Schallreuter et al., 1993). In contrast, HPS-1 caused by the homozygous mutation IVS17-2C>T resulted in death secondary to lung fibrosis and pulmonary insufficiency (Brantly et al., 2000), and the double homozygous mutation at amino acids S459 and S463 caused colitis with small bowel involvement in a patient at the age of 17 yr old (Hermos et al., 2002).
While HPS-1 heterozygotes are clinically asymptomatic, abnormalities can be detected on the cellular level. Individuals heterozygous for the mutation P324ins had decreased numbers of platelet dense granules detected by electron microscopy and decreased platelet aggregation in response to stimulation with various agonists (Gonzalez-Conejero et al., 2003).
Cell biology and biochemistry
HPS1 protein is reported to be ubiquitous and primarily localized to the cytosol, with a small proportion being membrane associated (Dell'Angelica et al., 2000a; Oh et al., 1996). Immunostaining for the HPS1 protein in normal fibroblasts was reported to yield a relatively homogeneous cytoplasmic pattern (Oh et al., 2000), while in normal melanocytes HPS1 demonstrated a perinuclear reticular pattern, and a more granular appearance (Oh et al., 2000; Richmond et al., 2005). In FME melanoma cells, endogenous HPS1 colocalized perinuclearly with both tyrosinase and tyrosinase related protein 2 (TRP2)/dopachrome tautomerase (DCT), melanogenic enzymes normally transported to the melanosome. No colocalization was noted with TGN46, a trans-Golgi marker, or lysosomal associated membrane protein-1 (LAMP-1), a marker of late endosomes and lysosomes (Oh et al., 2000). Ultrastructurally, HPS1 was found in melanoma cells on tubulovesicular structures near the Golgi apparatus, on uncoated vesicles and early melanosomes (Oh et al., 2000).
Subcellular fractionation studies demonstrated that in melanotic and non-melanotic cells, HPS1 protein was associated with the HPS4 protein in the ∼200 kDa cytosolic BLOC-3 complex, with a minor proportion of BLOC-3 also found to be membrane associated (Dell'Angelica et al., 2000a; Martina et al., 2003; Nazarian et al., 2003; Oh et al., 2000). In melanoma cells, HPS1 is also associated with large vesicle/organelle membranes consistent with melanosomes in a 500 kDa complex together with the HPS4 protein. The 500 kDa complex is also detected in nonmelanotic cells such as fibroblasts in low amounts associated with endosomal/vesicular cell fractions (Chiang et al., 2003; Oh et al., 2000). Interestingly, although the HPS1 and HPS4 proteins copurified by immunoprecipitation, gel filtration and sedimentation velocity analysis, no direct binding of HPS1 to HPS4 was detected by yeast two-hybrid analysis, suggesting that at least one other subunit is a component of BLOC-3 (Chiang et al., 2003; Martina et al., 2003; Nazarian et al., 2003; Oh et al., 2000).
Lack of endogenous HPS1 may affect membrane trafficking differently in fibroblasts compared with in melanocytes. One group noted that in immortalized fibroblasts from pale ear mice (the mouse model for HPS-1), LAMP-1 appeared more dispersed and less perinuclear in distribution compared with in control cells, and suggested that the HPS1 protein is required for clustering of endosome and lysosomes in the perinuclear region and that movement of those organelles is impaired in BLOC-3 deficient fibroblasts (Falcon-Perez et al., 2005; Nazarian et al., 2003). It was noted that LAMP-2 in HPS-1 fibroblasts also exhibited an abnormal dispersed distribution when compared with control cells, but the distribution of CD63/LAMP-3 was the same as in control fibroblasts (Dell'Angelica et al., 2000a). In contrast, in melanocytes from HPS-1 patients, CD63/LAMP-3 and LAMP-1 were found in large vesicular structures, compared with the more granular pattern seen in control melanocytes, but LAMP-2 distribution was normal (Richmond et al., 2005).
HPS1 appears to play a role in regulating protein traffic targeted to the melanosome. In melanocytes derived from HPS-1 patients, immunostaining for tyrosinase, Tyrp1 and TRP2/DCT produced a pattern with large vesicular structures in the cell body and dendrites (Boissy et al., 1998; Richmond et al., 2005), similar to that seen when HPS-1 melanocytes are stained for LAMP-1 and LAMP-3, instead of a small granular pattern seen throughout the control cells. The mislocalization of tyrosinase resulted in decreased melanin production (Boissy et al., 1998). On electron microscopic examination, cultured human HPS-1 melanocytes are observed to contain enlarged membrane bound compartments up to 2 μm in length containing 50 nm vesicles, melanosomes and other membranous structures (Boissy et al., 1998; Richmond et al., 2005); these compartments were also identified occasionally in 10% of melanocytes in skin biopsies from HPS patients and in melanoma cells in which HPS1 expression was abrogated by the introduction of anti-sense HPS1 cDNA (Sarangarajan et al., 2001). However, in another study, skin biopsies from 40 patients bearing the 16-bp duplication mutation did not detect any enlarged structures (Toro et al., 1999).
Recent data indicates that defects in HPS1 (and HPS4) also cause more global effects. Cultured cells defective in HPS1 and HPS4 have aggregated and distended Golgi apparati, longer dendrites and flatter cell bodies, suggesting a possible cytoskeletal defect (Chiang et al., 2005).
Mouse model
In mice, two allelic strains of pale ear (ep) have been sequenced at the Hps1 locus (Feng et al., 1997; Gardner et al., 1997) (Figure 1). The mouse Hps1 protein is predicted to be 703 amino acids and to have a molecular mass of 79.7 kDa. On the amino acid level, the mouse sequence is 81% identical to human HPS1 and 89% similar (Feng et al., 1997; Gardner et al., 1997). Mouse HPS1 and human HPS4 were co-purified together from mouse immortalized melanocytes expressing human HPS1, demonstrating the conservation of protein–protein interaction domains across species (Chiang et al., 2003). In platelets from pale ear mice, decreased dense granule contents serotonin, ATP and ADP were noted, and decreased numbers of dense granules were observed by electron microscopy (Novak et al., 1984; Swank et al., 1998).
Enlarged pigmented structures termed macromelanosomes were observed in the choroid of the eyes of pale ear mice and melanocytes cultured from the mice were also noted to have the macromelanosomes (Gardner et al., 1997). However, in vivo examination of dorsal back follicular melanocytes did not find similar enlarged structures (Nguyen et al., 2002).
While other HPS strains are relatively homogenously hypopigmented, the pale ear mouse and the model for HPS-4 (light ear mouse), are unique in having a coat color similar to the parental black-colored C57BL/6 strain, but in also having markedly hypopigmented ears, tails, and paws. Studies show that there is a decreased number of dermal melanocytes at these sites, suggesting that defects in HPS-1 (and HPS-4) may affect melanocyte migration to those areas during development or alternatively may affect melanocyte survival and/or proliferation (T. Nguyen and M.L. Wei, personal communication), consistent with the observations of altered cytoskeletal elements in HPS1 and HPS4 cells observed by others (Chiang et al., 2005).
HPS-2
Gene and protein structure
HPS-2 is caused by mutations in the AP3B1 gene encoding the β3A subunit of the heterotetrameric adapter protein complex AP-3. AP-3 plays a role in mediating cargo protein selection into transport vesicles and trafficking those membrane proteins to the lysosome (Bonifacino and Dell'Angelica, 1999). The AP-3 molecule consists of β3A-, δ-, μ3-, and σ3-subunits (Figure 2). Electron microscopic studies of a related molecule, the AP-2 adaptor, suggest that the β3A and δ-subunits of AP-3 have three domains: the head or core region, which mediates protein–protein interactions with the other subunits, the hydrophilic hinge region and the ear or appendage region (Heuser and Keen, 1988). The gene encoding β3A consists of 3968 bp comprising 27 exons (Huizing et al., 2002).

Mutations found in HPS-2 and pearl. Dell'Angelica (1999), Clark (2003), Huizing (2002), Feng 1999, 2000
Clinical features
Four patients have been reported in the literature with HPS-2 (Clark et al., 2003; Huizing et al., 2002) which is distinguished from the other forms of HPS by the presence of neutropenia and susceptibility to recurrent respiratory illnesses. An initial diagnosis of Chediak–Higashi Syndrome (CHS), a related disorder characterized by pigment dilution, recurrent infections and large intracellular granules, may initially be considered (Huizing et al., 2002); however, the large intracellular granules seen in CHS are not present in HPS-2.
Two brothers who were compound heterozygous with a 63 bp deletion at amino acid K389 and a single nucleotide substitution at L580R both had neutropenia, recurrent upper respiratory infections and otitis media, congenital hip dislocations secondary to dysplastic acetabulae and neurological abnormalities including poor balance. They also had dermatologic findings similar to those found in HPS-1 patients: acanthosis nigricans and hypertrichosis. Both also had slightly depressed results on pulmonary function testing and granulocyte hypoplasia on bone marrow biopsy (Shotelersuk et al., 2000). On the cellular level, these patients had detectable presence of the β3A protein, albeit in diminished amounts (Dell'Angelica et al., 1999).
A patient with two nonsense mutations at R509X and E659X had a very serious phenotype, which correlated with the lack of detectable β3A mRNA and protein in his cells. No μ3 protein was detected either, suggesting that in the absence of binding to β3A, the μ3 subunit was degraded. The patient had severe respiratory infections, leading to the need for continual oxygen supplementation, neutropenia requiring G-CSF administration, mild conductive hearing loss, hemorrhage requiring platelet transfusions and failure to thrive (Huizing et al., 2002).
A fourth patient reported with HPS-2 had the classic features of oculocutaneous albinism and platelet defects, and had immunodeficiency necessitating treatment with immunoglobulin and prophylactic antibiotics. On the cellular level, this patient had no detectable β3A, δ, or μ3A protein by immunoblot analysis, but did have residual σ3A protein (Clark et al., 2003).
Cell biology and biochemistry
AP-3 has been shown to mediate protein trafficking to the lysosome and lysosome-related compartments via binding to amino acid-encoded sorting signals in the cytoplasmic tails of lysosomally targeted molecules (Bonifacino and Traub, 2003; Ihrke et al., 2004; Sugita et al., 2002). Specifically, the amino acids tyrosine and leucine form the basis of two different sorting signals that have been demonstrated to bind to AP-3. In cells derived from HPS-2 patients deficient in AP-3, molecules normally targeted to lysosomes (e.g. CD63/LAMP-3, LAMP-1, LAMP-2, and CD1b), have an increased cell surface expression due to impaired trafficking to the lysosome, while the trafficking of the nonlysosomally targeted transferrin receptor (TfR) and cation-dependent mannose 6-phosphate receptor remained unchanged (Clark et al., 2003; Dell'Angelica et al., 1999; Sugita et al., 2002).
The misrouting of the lysosomally targeted molecules such as LAMP-1 does not appear to play a central role in HPS disease pathogenesis, however, since only those HPS mouse models that harbor mutations in the AP-3 molecule subunits (i.e. pearl and mocha mouse strains) have detectable increased cell surface expression of LAMP-1, whereas the other mouse models of HPS have normal cell surface levels (Dell'Angelica et al., 2000a). It is more likely that the LAMP-1 molecule serves as a marker for dysregulated membrane trafficking and the disease phenotype is a result of disrupted trafficking to LROs such as the melanosome, platelet dense granule, lytic granule, and lung lamellar body. For example, in cultured melanocytes derived from an HPS-2 patient, the tyrosinase protein (a melanogenic enzyme necessary for pigment synthesis) is mislocalized to a perinuclear compartment, compared with a perinuclear and dendritic distribution in control melanocytes (Huizing et al., 2001b). The steady state distribution of another melanosomal protein, Tyrp1, appeared unchanged by defects in AP-3 (Huizing et al., 2001b), suggesting that Tyrp1 primarily utilizes an AP-3 independent pathway to melanosomes, or that Tyrp1 normally uses an AP-3 pathway, but in the absence of AP-3 can access a second, AP-3-independent, pathway.
Recent evidence is consistent with AP-3 mediating a significant trafficking pathway from endosomes to lysosomes and LROs. In HepG2 cells, AP-3 is localized to endosomal structures that contained transferrin, TfR, asialoglyoprotein receptor, and the lysosome-associated membrane proteins LAMP-1 and 2 (Peden et al., 2004). In AP-3 deficient cells, molecules normally targeted to lysosomes appear to accumulate in endosomes and on the cell surface, and endosomes appear to increase in size. In HPS-2 lymphoblastoid cell lines, the CD1b molecule, normally targeted to lysosomes, colocalizes instead with TfR, a marker for early endosomes, as well as being increased on the cell surface (Sugita et al., 2002). In melanocytes from HPS-2 patients, abundant multivesicular structures resembling endosomes are seen, containing intralumenal tyrosinase enzyme activity, whereas similar structures were only occasionally seen in control melanocytes (Huizing et al., 2001b). In CTL from HPS-2 patients, an increased size of the entire endosomal network was observed (Clark et al., 2003). Together, these data suggest that molecules targeted to lysosomes or LROs first traverse from the TGN to an endosomal compartment where AP-3 mediates sorting and subsequent trafficking to the lysosome or LRO. In the absence of AP-3, molecules accumulate in the endosomal compartment, causing increased endosomal size, and are recycled to the cell surface by a default mechanism.
Investigations into the cellular basis for the immunodeficiency seen in HPS-2 patients demonstrated an impairment in the presentation of lipid antigen to T cells by CD1b molecules transduced into B-lymphoblastoid cells derived from an HPS-2 patient, whereas transduced CD1c molecules presented antigen at the same level as wild type cells (Sugita et al., 2002). HPS-2 CTL had a decreased ability to kill targets and to secrete the lysosomal hydrolase hexosaminidase with T-cell receptor linking. Incubation of HPS-2 CTL with target cells failed to induce polarization of lytic granules to the immunological synapse, suggesting that AP-3 function is required for microtubule-mediated movement of lytic granules (Clark et al., 2003).
The pigment dilution and neutropenia characterizing HPS-2 are similar to findings in a disease affecting dogs, canine cyclic neutropenia (also called gray collie syndrome), a stem-cell disease in which the number of neutrophils oscillates in weekly phases; the defective gene in canine cyclic neutropenia is also AP3B1 (Benson et al., 2003). In neutrophils, AP-3 was associated with the serine protease neutrophil elastase (NE) via a tyrosine based signal in the cytoplasmic tail of NE. AP-3 mediated trafficking of NE to lysosome-like granules (Benson et al., 2003), likely azurophilic granules (Bainton, 1999). In the absence of intact AP-3, NE accumulated at the plasma membrane. Furthermore, mutations in the NE molecule which disrupted the tyrosine based AP-3 recognition signal caused plasma membrane localization of NE and were associated with the human genetic disease severe congenital neutropenia (Horwitz et al., 2004). Thus, disruption of AP-3 mediated NE targeting to an LRO appears to underlie the neutropenia observed in HPS-2 and gray collie syndrome. The downstream events that lead to clinical neutropenia remain unknown.
Two syndromes similar to HPS-2 in affecting CTL granule mobility/function result in clinical signs of hemophagocytic lymphohistiocytosis (HLH). The Griscelli syndrome (caused by mutations in the RAB27A gene) and the Chediak–Higashi syndrome (caused by mutations in the LYST gene) are syndromes that also cause pigment dilution, platelet dysfunction and CTL granule defects; both also lead to HLH (Menasche et al., 2000; Nagle et al., 1996; Rubin et al., 1985). HLH without pigment dilution can also be caused by mutations in the perforin 1, syntaxin 11 or MUNC13-4 genes (Feldmann et al., 2003; zur Stadt et al., 2005; Stepp et al., 1999). An earlier study attributed the accumulation of ceroid material in HPS reticuloendothelial cells as secondary to hemophagocytosis or cellular ingestion of erythrocytes (White et al., 1973). It is not clear if perhaps the HPS-2 patients, aged 5, 20, and 25 yr at the time of reporting, may experience later onset of clinical HLH compared with in the other syndromes, or perhaps T-cell type-specific defects account for the clinical differences (i.e. distinct subsets of T cells or NK cells are affected by granule defects in the different syndromes).
Mouse model
The mouse model for HPS-2 is the pearl strain (pe), which exhibits pigment dilution, prolonged bleeding and impaired kidney and platelet lysosomal enzyme secretion (Swank et al., 1998). This strain also exhibits reduced sensitivity in the dark-adapted state and is suggested to also be a model for human congenital stationary night blindness (Balkema et al., 1983). Constitutive secretion of lysosomal enzymes from the kidney proximal tubules is decreased to one-third of normal (Novak and Swank, 1979) and thrombin mediated secretion of lysosomal enzymes from platelets is 50% of normal (Novak et al., 1984). Enlarged multilamellar structures resembling lysosomes are observed in kidney proximal tubule cells from pearl mice and cultured pearl melanocytes have distinctive organelles with multilamellar concentric internal membranes, also thought to be lysosomes (Zhen et al., 1999). The prolonged bleeding in pearl mice can be corrected by bone marrow transplantation, indicating that the AP-3 defect affects hematopoietic progenitor cells (McGarry et al., 1986), in agreement with the observations in the gray collie syndrome and congenital neutropenia.
Three mutant alleles of pearl have been described: pe, pe8J and pe9J (Feng et al., 1999, 2000). In the cases of pe and pe8J, residual decreased amounts of mRNA are detected (Feng et al., 1999; Yang et al., 2000; Zhen et al., 1999). Low levels of β3A protein are expressed in pe, and small amounts of AP-3 heterotetramers are detected by immunoprecipitation from extracts of pe spleen cells, formed by the association of the truncated β3A subunit with the remaining δ-, μ- and σ-subunits (Peden et al., 2002). Transgenic mice in which the Ap3b1 gene has been disrupted to produce a null phenotype have a greater degree of coat color pigment dilution compared with pe mice and cell extracts from the transgenic mice have a decreased of detection of δ, μ, and σ due to degradation of the unassembled subunits (Yang et al., 2000). Thus pe mice are phenotypic hypomorphs.
Immunofluorescence analysis of fibroblasts from pearl and Ap3b1 null mice demonstrate increased cell surface expression of LAMP-1 and LAMP-2, in agreement with studies on cells from HPS-2 patients, and steady-state tyrosinase was mislocalized in Ap3b1-null melanocytes (Dell'Angelica et al., 2000a; Yang et al., 2000).
A related mouse strain is the mocha mouse (mh), in which a different subunit of AP-3 is defective, the δ-subunit; no corresponding patients have been identified. There are two allelic strains of mocha, mh and mh2J, the first of which is a functional null and the second of which is a hypomorph (Kantheti et al., 1998, 2003). While the mocha strains have variable degrees of pigment dilution and platelet dysfunction, the most striking defects are neurological (Kantheti et al., 1998, 2003). In contrast, the pearl mice and HPS-2 patients (with defects in AP-3 subunit β3A) do not exhibit neurological deficits because a neuronal tissue-specific isoform of β exists, called β3B (Newman et al., 1995; Simpson et al., 1997). This neuronal isoform is intact in pearl mice (Zhen et al., 1999) and continues to mediate normal neuronal AP-3 functions such as synaptic vesicle formation from endosomes (Faundez et al., 1998; Seong et al., 2005), despite defects in the non-neuronal isoform.
In contrast, no tissue-specific forms of the δ-subunit have been detected. In mh mice a large out-of-frame deletion causes the absence of any detectable δ-protein in all tissue and cells tested, including brain; loss of δ also caused the loss of detection of the other 3 AP-3 subunits, due to instability of the unassembled units (Kantheti et al., 1998). The neurological defects in mocha mice were associated with a loss of immunoreactivity for the zinc transporter protein ZnT-3 and with a dramatic of reduction of vesicular zinc staining in the central nervous system, but with normal number and appearance of the normally zinc containing presynaptic vesicles (Kantheti et al., 1998). The ZnT-3 molecule has both tyrosine- and dileucine-based sequences (Kantheti et al., 1998), consistent with a role for AP-3 in linking the ZnT-3 cargo molecule via its cytoplasmic targeting signals with vesicular coat proteins, such as clathrin, for trafficking to target organelles.
HPS-3
Gene and protein structure
The HPS3 gene is found on chromosome 3q24, has 17 exons with a 3015 open reading frame encoding a protein of 1004 amino acids predicted to have a molecular mass of 113.7 kDa (Anikster et al., 2001), but with an apparent molecular mass of 130 kDa by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis (Nazarian et al., 2003). A clathrin binding site is predicted from codons 172–176 (Figure 3). The mRNA transcript is 4.4 kb and appears to be ubiquitous, expressed in heart, brain, placenta, lung, liver, skeletal muscle, kidney, and pancreas (Anikster et al., 2001).

Mutations found in HPS-3 and cocoa. Anikster (2001), Boissy (2005), Huizing (2001a,b), Suzuki (2001)
Twenty-one patients with HPS3 have been reported in the literature, with eight mutations described (Anikster et al., 2001; Huizing et al., 2001a). The most common mutation is found among families from central Puerto Rico due to a founder effect; all of these patients are homozygous for a large 3904-bp deletion encompassing all of exon 1, >2 kb of upstream sequence and 673 bp of intron 1 (Figure 3). One mutation is associated with patients of Ashkenazi Jewish descent: the IVS5 + 1G>A splice site mutation occurred in five Ashkenazi Jews, and not in any of the three patients not of Ashkenazi Jewish descent (Huizing et al., 2001a).
Clinical features
HPS-3 is clinically mild compared with HPS-1 (Anikster et al., 2001; Huizing et al., 2001a; Tsilou et al., 2004). In some patients, extra-ocular symptoms were so mild, they carried an initial diagnosis of ocular albinism for decades. On average, the best corrected is 20/125 (range 20/50–20/320) compared with 20/160 in HPS-1, and the iris transillumination in HPS-3 on average is less than in HPS-1. All reported HPS-3 patients exhibited excessive bruising and epistaxis, but only an occasional patient required transfusion. Pulmonary testing revealed minimal reduction in FVC (75–96% of normal).
Cell biology and biochemistry
Studies of HeLa cells demonstrated that the HPS3 protein associated with the HPS5 and HPS6 proteins in a multimeric protein complex, BLOC-2, which had an estimated molecular mass of 340 kDa (Di Pietro et al., 2004).
Immunofluorescent imaging of melanocytes derived from HPS-3 patients demonstrated that molecules normally targeted to later stage melanosomes (e.g. the melanogenic enzymes tyrosinase and Tyrp1) were mislocalized (Boissy et al., 2005; Richmond et al., 2005); mislocalization of tyrosinase correlated with decreased levels of melanin in melanocytes (Boissy et al., 2005). In contrast, the steady-state distribution of molecules targeted to stage I melanosomes (e.g. silver/Pmel17/gp100 and melan-a/MART1), were found to be unchanged from that in control cells. Transmission electron microscopic analysis of HPS-3 melanocytes detected 50-nm vesicles, containing melanogenic enzymatic activity as measured by DOPA staining, more numerously and in a more dispersed distribution than found in control melanocytes. The DOPA staining also detected melanogenic enzymatic activity in melanosomes, suggesting that melanogenic enzymes can access melanosomes via an HPS3-independent pathway, and that the lack of detected melanin in later stage melanosomes in HPS-3 cells is due to the limiting quantity of another (presumably mis-trafficked) molecule. Studies have shown that the activity of tyrosinase increases with phosphorylation and upon association with Tyrp1 (Wu and Park, 2003; Park et al., 1993, 1999), and that tyrosinase phosphorylation may be RACK1 mediated (Park et al., 2004); thus molecular events other than delivery of melanogenic enzymes to melanosomes influence melanin production. The LAMP-1 and LAMP-3 molecules had a similar pattern of mislocalization in HPS-3 melanocytes as did tyrosinase: in HPS-3 cells, the distribution was more diffuse than in control cells (Boissy et al., 2005).
HPS3 has a clathrin binding motif (Anikster et al., 2001). Clathrin was co-immunopreciptated with HPS3, clathrin and HPS3 were observed to partially colocalize and live cell imaging demonstrated fusion of HPS3 labeled vesicles with clathrin labelled vesicles. Furthermore, mutation of the clathrin binding motif in HPS3 abrogated the association of HPS3 with small vesicles and resulted in a primarily cytoplasmic localization for HPS3, suggesting a functional role for clathrin in localizing HPS3 to vesicular membranes (Helip-Wooley et al., 2005).
Mouse model
The cocoa mouse strain (coa) is the model for HPS-3 (Novak et al., 1988; Suzuki et al., 2001). The mouse Hps3 gene contains 17 exons, coding for a protein with 1002 amino acids and a predicted molecular mass 113.1 kDa, which appears to be ubiquitously expressed (Suzuki et al., 2001). Four allelic strains of cocoa have mutations in the Hps3 gene (Figure 3).
Cocoa is distinguished from most of the other HPS mouse models by normal secretion of kidney lysosomal enzymes (Novak et al., 1988; Swank et al., 1998) and skin fibroblasts from cocoa mice do not have a defect in basal levels of secretion of lysosomal enzymes (Di Pietro et al., 2004).
Immortalized cultured melanocytes from cocoa mice show little visible pigment and melanin production was decreased to approxmately 25% of that found in control cells (Suzuki et al., 2001). Ultrastructural studies of cocoa eye tissue found decreased numbers of melanosomes in the retinal pigment epithelium and choroid; in addition, multilamellar bodies and abnormal melanosomes were observed in the choroid (Suzuki et al., 2001). Examination of the dorsal back follicular melanocytes by transmission electron microscopy showed a preponderance of immature melanosomes, with an apparent block in melanosome biogenesis occurring before the formation of stage II melanosomes (Nguyen et al., 2002).
The association of Hps3, Hps5, and Hps6 in the BLOC-2 complex was demonstrated by coimmunoprecipitation, but yeast two-hybrid analysis did not detect direct binding of Hsp3 to Hps5 or Hps6, suggesting the possible existence of another small subunit (Gautam et al., 2004; Zhang et al., 2003).
HPS-4
Gene and protein structure
The HPS4 gene is found on chromosome 22q11.2-q12.2 and has a distant homology with the yeast CCZ1 protein (Anderson et al., 2003; Hoffman-Sommer et al., 2005) which is implicated in trafficking and fusion of vesicles from the yeast prevaculoar compartment (functionally equivalent to the mammalian late endosome) to the vacuole (equivalent to the mammalian lysosome). HPS4 consists of 14 exons that covers approximately 32 kb of genomic DNA (Anderson et al., 2003). A major transcript consisting of 14 exons with a 2127-bp open reading frame, coding for a 708-amino acid protein (predicted molecular mass of 76.9 kD, apparent molecular mass of 100 kDa by SDS-PAGE (Martina et al., 2003; Nazarian et al., 2003)), is expressed in all tissue types tested. A second alternatively spliced isoform has 12 exons with a 2112-bp open reading frame, coding for a 703-amino acid protein, is limited in expression to brain, thymus, lymph node, fetal liver, bone marrow and melanocytes and no homologs have been identified in other mammalian species for this minor protein. These two transcripts differ in 5’ exon usage, but are identical after codon 15 in the major isoform (codon 10 of the minor isoform). Three other transcript variants of uncertain biological importance have also been reported (Anderson et al., 2003).
To date, 15 patients worldwide have been reported with HPS-4 and 10 mutations in the HPS4 gene have been identified (Figure 4). The functional importance of the far carboxyl terminus of HPS4 is demonstrated by homozygous frameshift mutations found in two patients, one patient carrying a deletion at codon 685, resulting in a loss of 24 normally coded amino acids from the carboxyl terminus and another carrying an insertion of five nucleotides at codon 698, causing a loss of 10 normally coded amino acids. The first patient had severe, progressive pulmonary fibrosis notable for hypoxemic respiratory failure requiring supplemental oxygen (Bachli et al., 2004). The second patient, in which HPS4 RNA was detected in fibroblasts, suffered from severe pulmonary fibrosis resulting in death at 61 yr of age (Anderson et al., 2003).

Mutations found in HPS-4 and light ear. Suzuki (2002), Anderson (2003), Bachli 2004
Eight polymorphisms of HPS4 have been detected, four of which cause amino acid substitutions: E229G, V552M, H606Y, and Q625H (Anderson et al., 2003).
Clinical features
As in other subtypes of HPS, HPS-4 patients have great variability in the degree of hypopigmentation. Many, but not all, have clinical signs and symptoms of platelet dysfunction, marked by epistaxis, bruising and in the case of female patients, menorrhagia. Best corrected visual acuity varies from 10/30 to 20/200 (Anderson et al., 2003). HPS-4 patients can have a severe clinical phenotype, with features similar to those observed in HPS-1 patients. In one study, one of seven patients with HPS-4 was diagnosed with granulomatous colitis, and three of seven had restrictive pulmonary fibrosis (Anderson et al., 2003). In another detailed clinical study of the patient carrying the homozygous P685del mutation who suffered from severe pulmonary fibrosis, skin biopsy was reported to show normal numbers of melanocytes and decreased melanin content by Masson–Fontana staining and perivascular macrophages containing ceroid pigment; lung biopsy revealed an increased number of type II pneumocytes with foamy cytoplasm, and macrophages contained ceroid pigment (Bachli et al., 2004).
Cell biology
Platelets from an HPS-4 patient were found to have altered localization of the ATP-dependent pump MRP4 (also known as ABCC4), a transport pump for cyclic nucleotides and nucleotide analogs (Jedlitschky et al., 2004). In control platelets, MRP4 is found in intracellular granules, as well as on the plasma membrane, whereas in HPS-4 platelets, intracellular staining was greatly decreased, with staining predominantly seen on the cell surface.
Mouse model
The corresponding mouse model for HPS-4 is the light ear (le) strain (Suzuki et al., 2002). The mouse light ear protein is predicted to have 671 amino acids and have a molecular mass of 72.7 kD. Two isoforms of RNA transcripts are detectable in light ear tissue, 3.6 and 3.1 kb in size. Ultrastructural analysis of melanosomes in the eyes of light ear mice showed abnormalities in the retinal pigment epithelium, in which melanosomes were decreased in number, and in the choroid, which had markedly enlarged melanosomes. Cultured melanocytes from light ear skin had small, abnormally shaped immature melanosomes (Suzuki et al., 2002), consistent with observations in vivo on epidermal melanocytes from light ear tail skin (T. Nguyen and M.L. Wei, personal communications).
In a cell line established from light ear mouse skin, cellular activity of lysosomal hydrolases were elevated compared with in control cells, while secreted lysosomal hydrolase activities were 10% of control cell levels (Delprato et al., 2000). This result was consistent with the earlier findings of increased kidney lysosomal hydrolase activity in light ear and pale ear mice and concomitant decreased levels of activity found secreted into urine (Meisler, 1978; Novak and Swank, 1979), suggesting impaired secretion.
HPS-5
Gene and protein structure
The HPS5 gene is found on chromosome 11p14, has 23 exons and encodes a protein 1129 amino acids in length with a calculated molecular mass of 127 kDa (Huizing et al., 2004; Zhang et al., 2003) and an apparent molecular mass of 170 kDa (Di Pietro et al., 2004). The 4.8 kb transcript was found to be expressed in all tissues tested, but was very highly expressed in lung and testis (Huizing et al., 2004). Two alternatively spliced isoforms were detected, both predicted to result in proteins with 114 fewer amino acids at the amino terminus, because of an alternative start sites in exon 5 (Huizing et al., 2004; Zhang et al., 2003). Both of these latter isoforms were also expressed in all tissues surveyed, but one (719 bp) was expressed at high levels in placenta, kidney, testis, and ovary, while the other (588 bp) was found to be expressed at high levels in placenta, lung, and thymus (Huizing et al., 2004).
Seven disease causing mutations in HPS5 have been detected (Huizing et al., 2004; Zhang et al., 2003) (Figure 5). A pair of siblings had two missense mutations (L624R and T1098I) that both occurred together in the homozygous state, so whether both mutations are disease causing or only one is, with the other change being a polymorphism, could not be determined (Huizing et al., 2004).

Mutations found in HPS-5 and ruby eye-2. Huizing (2004), Zhang 2003
Clinical features
The clinical features of five individuals with HPS-5, ages 3–51 yr, have been reported (Huizing et al., 2004; Zhang et al., 2003). Most patients were of northern European descent; one was Turkish. All had nystagmus or visual symptoms (such as poor tracking) that led to an early diagnosis of albinism. Visual acuity ranged from 20/100 to 20/200. All had bruising; one patient had menorrhagia and metrorrhagia requiring blood or platelet transfusions. All had absent platelet dense bodies, leading to a diagnosis of HPS. None had a diagnosis of inflammatory bowel disease and none had shortness of breath. Minimal to no impairment was detected by pulmonary function testing (average FVC was 96% of normal, range was 80–111%). Four of five patients had mildly to minimally elevated creatinine clearance, testing for the fifth patient was not noted. All patients had elevated cholesterol levels, and a few had mildly elevated triglycerides as well. The significance of these elevated lipid levels, and whether they are a result of the underlying membrane trafficking defect, is not known.
Cell biology and biochemistry
As noted previously, HPS3, HPS5, and HPS6 associate together in a complex designated BLOC-2 (Di Pietro et al., 2004). Studies on fibroblasts from HPS-5 patients noted that the distribution of LAMP-3 (CD63) was confined to granules in a perinuclear region, unlike in normal fibroblasts, in which LAMP-3 was also found in granules distributed in the dendritic processes (Huizing et al., 2004). The HPS5 molecule was reported to bind to the cytoplasmic domain of the α3A integrin molecule (Wixler et al., 1999), but another group was unable to duplicate the finding (Zhang et al., 2003).
Mouse model
The ruby eye-2 (ru2) mouse strain is the model for HPS-5 (Zhang et al., 2003). The mouse Hps5 gene is on chromosome 7 and has a 3381-bp open reading frame with 23 exons, encoding a 1126 amino acid protein, 81% homologous to the human sequence, and with predicted molecular mass 126.3 kDa. By Northern blot analysis, all tissues examined contained the 4.8 kb transcript. Alternate transcripts were also detected in kidney tissue, one that results in truncation of 165 amino acids from the amino terminus, and another that truncates 600 amino acids from the carboxyl terminus. The significance and general tissue distribution of these transcripts is not known.
The melanosome morphology in ruby eye-2 is abnormal in eye and skin. In retinal pigment epithelium, melanosomes are decreased in number and aberrantly shaped. In the choroid, there is also a decreased number, and several multi-melanosomal bodies are observed, a finding unique to ruby eye-2 and ruby (Zhang et al., 2003). In dorsal skin follicular melanocytes, the steady state distribution of melanosomes was shifted to more immature stages; most remained spherical, without progression to the elliptical shape characteristic of stage II melanosomes (Nguyen et al., 2002), suggesting that the ru2 protein functions in melanosome maturation from stage I to stage II.
In COS7 cells expressing introduced Hps5 and Hps6, the two molecules coimmunoprecipitated, demonstrating that, similar to the human counterparts, these proteins associated together in a complex. Yeast two-hybrid analysis demonstrated a direct interaction between Hps5 and Hps6 (Zhang et al., 2003).
HPS-6
Gene and protein structure
The HPS6 gene, on chromosome 10q24.32, consists of a single exon (Zhang et al., 2003). The predicted HPS6 protein length is 775 amino acids (Figure 6).

Mutations found in HPS-6 and ruby eye. Hess (2003), Zhang (2003)
Clinical features
Three patients have been reported in the literature with HPS-6 (Zhang et al., 2003). One 39-yr-old woman had no pulmonary or gastrointestinal symptoms, but did have epistaxis and bleeding after dental extractions and surgeries. Two other patients were both under 27 yr of age and had typical oculocutaneous albinism but no pulmonary fibrosis or colitis.
Mouse model
The mouse model for HPS-6 is the ruby eye (ru) strain (Zhang et al., 2003). The Hps6 gene is on chromosome 19 and encodes a 2418-bp open reading frame, and is similar to the human gene, containing a single exon. The encoded 805 amino acid protein has a predicted molecular mass of 88.8 kDa and has 80% identity with the human HPS6 protein; the mouse and rat proteins have an extension of 31 amino acids.
There are four allelic ruby eye strains (Zhang et al., 2003). Two strains, ru and ru4J, have in-frame deletions in Hps6, a three amino acid deletion encompassing amino acids H187 to P189 and a 22 amino acid deletion encompassing amino acids L65–W86, respectively (Figure 6). The loss of the three amino acids in the ru strain suggested that the interaction of Hps6 with Hsp5 might depend on these three residues (his/cys/pro). This was confirmed using yeast two hybrid analysis which found that Hps6ru does not bind to Hps5wt (Gautam et al., 2004). When each residue in this binding sequence was singly mutated to an alanine, binding between Hps5 and Hps6 remained intact, suggesting that the sequence only indirectly mediates the protein–protein interaction, perhaps by influencing secondary structural features.
In ruby eye and ruby eye-2 mice, as with the majority of HPS mouse strains (Swank et al., 1998), there is a decreased rate of kidney lysosomal enzyme secretion after testosterone treatment, compared with in control cells (Novak et al., 1980). However, ruby eye skin fibroblasts demonstrate no defect in basal secretion of lysosomal enzymes (Di Pietro et al., 2004) suggesting that perhaps in the kidney, an LRO (vs. the lysosome) is affected by mutations in HPS genes. In mast cells collected from ruby eye mice, an approximately threefold increase in the number and duration of transient fusion events with the plasma membrane was recorded (Oberhauser and Fernandez, 1996), suggesting that the Hps6 protein may have a role in mediating the exocytosis of mast cell granules. However, this function in exocytosis could not be assigned unambiguously to Hps6, as cells derived from control mice with the same genetic background as the ruby eye mice could not be assayed.
HPS-7
Gene and protein structure
The DTNBP1 gene is defective in HPS-7, encodes the dysbindin protein on chromosome 6p22.3 and has 10 exons (Li et al., 2003; Straub et al., 2002). Six non-disease causing polymorphisms have been detected, and a single mutation has been reported (Figure 7).

(A) Mutations found in HPS-7 and sandy; (B) mutations found in HPS-8 and reduced pigmentation. (Gwynn 2004; Starcevic 2004)
Clinical features
The single patient reported with HPS-7, a 48-yr-old Portuguese woman, was homozygous for the Q103X mutation (Li et al., 2003). She had oculocutaneous albinism, easy bruisability, a bleeding tendency, mild shortness of breath with exertion, decreased lung compliance, but otherwise normal pulmonary function tests and high-resolution chest CT. No muscle weakness, ataxia or symptoms of schizophrenia were noted (Li et al., 2003, 2004).
Cell biology and biochemistry
In addition to a role in causing HPS-7, dysbindin is implicated in the molecular pathology of Duchenne muscular dystrophy (DMD) because of its binding to members of the dystrophin-associated protein complex, α- and β-dystrobrevin; the DMD gene encodes dystrophin (Benson et al., 2001). Yeast two-hybrid analysis also detected binding to a novel binding partner, myospryn, which associated with dysbindin via the coiled coil domain of dysbindin (Benson et al., 2004). The tissue expression of myospryn is limited to skeletal muscle and heart, and provides an example of a tissue-specific binding partner for the ubiquitously expressed dysbindin.
Genetic susceptibility to schizophrenia has been mapped to the DTNBP1 gene locus (Straub et al., 2002) and dysbindin appears to play a role in the exocytosis of glutamate containing synaptic vesicles in neurons. Overexpression of dysbindin in cultured primary cortical neurons induced the expression of SNAP25, a soluble protein that regulates membrane fusion events, and also upregulated synapsin I, a cytoskeletal protein associated with synaptic vesicles. Depression of intracellular levels of dysbindin mediated by siRNA resulted in decreased detection of SNAP25 and synapsin I by immunoblotting and reduced glutamate release by siRNA treated cells (Numakawa et al., 2004).
Mouse model
The mouse model for HPS-7 is the sandy (sdy) strain (Li et al., 2003; Swank et al., 1991). The defective gene, Dtnbp1, encodes a 51 kDa, 352 amino acid protein (Li et al., 2003) predicted to have a coiled-coil region between amino acids 88 and 177 (Benson et al., 2001).
In sandy mice, an inframe deletion from genomic nucleotides 3701–12 377 caused deletion of 52 residues (amino acids 119–172) comprising exons 6 and 7 and including the majority of the predicted coiled coil region (Figure 7). While in wild-type mice, a 1.65-bp transcript is expressed in brain, kidney, and heart (suggesting ubiquitous expression), in sandy mice a smaller 1.5-kb transcript is detected, and no protein product is detected by immunoblotting (Li et al., 2003).
In yeast two-hybrid analysis, dysbindin was found to bind β-dystrobrevin, in agreement with previous studies, and also to two other proteins, muted and pallidin, named for the HPS mouse strains in which they are defective, muted and pallid (Benson et al., 2001; Li et al., 2003). Muted and pallidin were previously shown to associate together in the multiprotein BLOC-1 complex, which included two other HPS proteins, rp and cappuccino (Ciciotte et al., 2003; Falcon-Perez et al., 2002; Gwynn et al., 2004).
Dysbindin was confirmed to be a component of BLOC-1 by coimmunoprecipitation (Li et al., 2003); size exclusion chromatography and sedimentation velocity analysis further confirmed that endogenous dysbindin and pallidin associated together in a 230 kDa complex (Falcon-Perez et al., 2002). The bulk of β-dystrobrevin did not appear to co-purify with BLOC-1 components, although a small amount may be associated with murine BLOC-1 (Li et al., 2003); no α- or β-dystrobrevin was isolated in bovine BLOC-1 (Starcevic and Dell'Angelica, 2004). Thus it is not clear if the α- and β-dystrobrevin proteins are involved in vesicle trafficking, or if dysbindin is a component of two separate multiprotein complexes which mediate distinct functions (Li et al., 2003). The β-dystrobrevin containing dystrophin-associated protein complex has been reported to interact with actin, as has pallidin (Blake et al., 2002; Falcon-Perez et al., 2002), but the functional significance of these interactions is unclear.
Evidence suggests that BLOC-1 has a role in membrane trafficking. Pallidin binds syntaxin 13, a member of the SNARE family of proteins that have a role in vesicle targeting, docking and fusion with target membranes (Advani et al., 1998). Syntaxin 13 has been localized to early and recycling endosomes, and also to endosomal vacuoles, where it is often found in clathrin-coated membrane areas (Prekeris et al., 1998). An additional member of the BLOC-1 complex is snapin, a protein previously implicated in the regulation of membrane fusion events (Ilardi et al., 1999; Buxton et al., 2003) Together with snapin, dysbindin, pallidin, muted, rp and cappuccino, two novel proteins, BLOS-1 and BLOS-2, are also components of BLOC-1 (Starcevic and Dell'Angelica, 2004). While all of the BLOC-1 subunits except for rp contain predicted coiled coil regions (implicated in mediating protein–protein interactions), non-coiled coil regions also appear capable of mediating protein–protein interactions, as deletion of the BLOS-1 coiled coil region is not required for BLOS-1 binding to pallidin (Starcevic and Dell'Angelica, 2004).
Melanosomes in sandy mice are markedly abnormal. In the eye, the retinal pigment epithelial melanosomes are decreased in number and the choroidal melanosomes are also decreased in number and smaller and irregularly shaped (Li et al., 2003). In the dorsal back skin follicular melanocytes, melanosome biogenesis was blocked at or before stage I and numerous aberrant vacuolar forms were noted (Nguyen and Wei, 2004).
HPS-8
Gene and protein structure
HPS-8 has been identified as due to a defect in the BLOC1S3 gene and is orthologous to the mouse rp gene. A large consanguineous family with HPS was identified to have a homozygous germline frameshift mutation (Morgan et al., 2006) (Figure 7).
Clinical features
Patients had incomplete oculocutaneous albinism and mild platelet dysfunction with easy bruising, hematomas after venesection, frequent epistaxis and prolonged bleeding after surgery or childbearing, sometimes requiring blood transfusion (Morgan et al., 2006).
Mouse model
The reduced pigmentation mouse was observed to have pigment dilution, increased kidney lysosomal glycosidase activities, increased bleeding times, decreased platelet dense bodies, predominantly immature melanosomes, decreased melanin levels and an abnormal intracellular tyrosinase distribution, findings consistent with being a model for an HPS subtype (Gibb et al., 1981; Gwynn et al., 2004; Nguyen et al., 2002; Swank et al., 1998).
The mouse gene is found on chromosome 7, consists of a 1867 bp ORF containing two exons encoding an 195 aa protein with an apparent molecular mass of ∼32 kDa (Gwynn et al., 2004; Starcevic and Dell'Angelica, 2004). Co-immunopreciptation studies, sedimentation analyses, yeast two-hybrid studies and size exclusion chromatography analyzed by immunoblotting established the protein defective in rp mice (BLOS3/rp), as a component of BLOC-1 when in the phosphorylated form (Gwynn et al., 2004; Starcevic and Dell'Angelica, 2004). However, unlike defects in other BLOC-1 components, the defect in the rp protein does not cause disruption of the BLOC-1 complex, and thus the phenotype is that of a hypomorph (Starcevic and Dell'Angelica, 2004).
Discussion
Hermansky–Pudlak syndrome is an example of a genetically heterogeneous syndrome that is the result of defects in protein trafficking along the endocytic/lysosomal pathway. HPS gene products can be divided into two types. Group A (AP3B1, AP3D1, RABGGTA, and VPS33A), are those that have homologues in yeast, which regulate trafficking to the yeast vacuole and in higher eukaryotes function as major regulators of trafficking to the lysosome. In contrast, the remaining HPS group B gene products (HPS1, HPS3, HPS4, HPS5, HPS6, DTNBP1, HPS8, Pldn, cno, and mu), are only found in metazoans (Li et al., 2004), and although containing regions with some homology to yeast proteins (Hoffman-Sommer et al., 2005), they have apparently developed as specialized cell types evolved with the need to form specialized organelles. This evolutionary development, together with the preponderance of data accumulating on the cellular effects of defects in HPS proteins, suggests that the HPS group B proteins function primarily to regulate membrane and protein trafficking to the ‘newer’ specialized organelles, the LROs, but may also contribute to lysosomal trafficking.
Mechanisms for cell type-specific disease expression
One intriguing aspect of HPS has been that the disease expression seems to be limited to a certain few specialized cell types despite ubiquitous tissue expression of HPS proteins. One explanation could lie in the way that different cell types developed pathways of biogenesis for their specialized organelles. In some specialized cell types, such as the cytotoxic T cells (CTL), the specialized organelle (in this case the lytic granule), is a secretory lysosome; i.e. there are no separate lysosomes distinct from the lytic granule (Stinchcombe and Griffiths, 1999). In contrast, in another specialized cell type, the melanocyte, studies suggested that the specialized organelle (the melanosome) coexists in the cell with lysosomes (Raposo et al., 2001). This may have implications regarding the impact of defects in HPS group A vs. group B proteins. Group B proteins might not affect cells with secretory lysosomes but rather primarily affect cells which had specialized LRO organelles that were distinct from lysosomes. Consistent with this prediction is the finding that the function of CTLs deficient in BLOC-1, -2, and -3 was found to be normal (Bossi et al., 2005).
Another potential mechanism for cell type specificity is illustrated in the case of the protein affected in HPS-7, dysbindin, which is observed to have a tissue specific protein binding partner, myospryn. So while the HPS proteins are ubiquitous, it is likely that HPS proteins function via binding to cell-type specific effectors (e.g. myospryn) and/or tissue-specific cargo such as tyrosinase (found only in pigment cells). Similarly, in the case of AP-3, the defective complex in HPS-2, tissue specific neuronal subunits provide a mechanism for mediating brain specific functions such as synaptic vesicle biogenesis. An additional mechanism contributing to cell-type specificity may come from regulating differential expression levels of HPS proteins in different cells types. For example, a particular antibody was able to detect endogenous HPS1 protein in human melanoma cells by immunoblotting and immunoprecititation, but was unable to detect endogenous HPS1 protein in HeLa cells by the same methods (Dell'Angelica et al., 2000a; Sarangarajan et al., 2001), suggesting the possibility that pigment cells may express higher levels of HPS1 compared with non-pigment cells. Further tissue specific regulation may derive from cell type-specific use of alternative transcripts; for example, alternatively spliced isoforms of HPS5 and HPS4 are expressed at different levels in different tissues (Anderson et al., 2003; Huizing et al., 2004).
Molecular pathology and HPS clinical subtypes
While all HPS patients suffer from oculocutaneous albinism (OCA) and prolonged bleeding, different subtypes, some with distinguishing features, are now recognized. Recent genetic and biochemical characterizations of HPS subtypes have enabled the classification of the subtypes into groups (Table 3); the heterogeneous clinical phenotypes of HPS can now be understood in the context of the molecular pathology. HPS-1 and HPS-4 subtypes are similar in causing the most serious morbidity and premature mortality, with affected individuals at high risk for developing restrictive pulmonary disease and inflammatory bowel disease. These clinical similarities reflect the molecular association of HPS1 and HPS4 to form the intracellular BLOC-3 complex; this complex regulates the biogenesis and/or function of the lung lamellar body as well as the platelet dense body and the melanosome. HPS-2 is unique in causing immunodeficiency. The HPS-3, HPS-5, and HPS-6 subtypes are clinically similar; the respective proteins associate to form the BLOC-2 complex, and defects in these proteins result in relatively mild symptoms of platelet dysfunction, without pulmonary involvement. Patients with HPS-7 and HPS-8, due to defects in BLOC-1 subunits, had moderate signs and symptoms of HPS: OCA, a bleeding tendency and mild pulmonary symptoms (in the case of HPS-7). The relatively mild systemic symptoms of HPS-7 and HPSS-8 contrast with the markedly severe pigmentary phenotype observed in the mouse strains deficient in BLOC-1 genes; however, the moderate phenotype of the few reported patients affected by defects in BLOC-1 may not be representative of BLOC-1 patients in general.
| HPS subtype | Protein complex | Clinical manifestations |
|---|---|---|
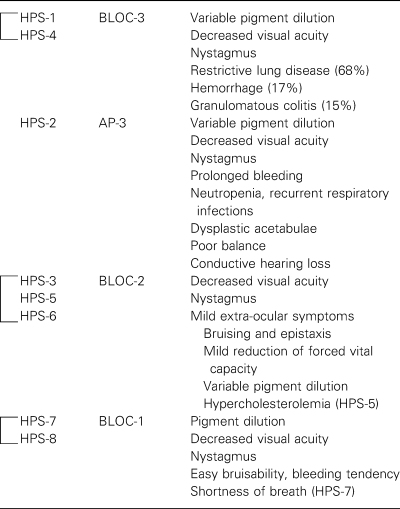 |
||
The pigment dilution in HPS appears to result from mistrafficking of melanogenic enzymes such as tyrosinase, which leads to decreased melanin production. The trafficking of additional other melanocyte factors may also be affected, as melanosome biogenesis appears to be blocked at immature stages. The bleeding diatheses appear to be secondary to mistrafficking of the MRP4/ABCC4 transporter in platelets with a consequent lack of intralumenal vesicular loading of nucleotides, leading to decreased secretion of these nucleotides with platelet aggregation and impaired clotting. Pulmonary fibrosis appears to result from impaired secretion of surfactant and phospholipids from abnormal appearing lung lamellar bodies. Moreover, congenital neutropenia results from mistrafficking of NE. Thus, defects in protein trafficking pathways and organelle function account for much of the pathophysiology seen in HPS (Table 4).
| Organelle | Cell type | Mistrafficked protein(s) | HPS subtype |
|---|---|---|---|
| Melanosome | Pigment cella | Tyrosinase | HPS-1–7 |
| Tyrp1, DCT/TRP2, CD63 | HPS-1, HPS-3 | ||
| Dense granule | Platelet | MRP4/ABCC4, CD63 | HPS-4 |
| Lytic granule | Cytotoxic T cell | CD63 | HPS-2 |
| Azurophilic granule | Neutrophil | Neutrophil elastase | HPS-2 |
| Lamellar body | Lung type II epithelial cell | ? | HPS-1, HPS-4 |
- aMelanocytes and retinal pigment epithelial cells.
HPS and melanocyte protein trafficking
Studies of HPS have also added to the understanding of critical aspects of melanocytes function. In humans and mice, the two major forms of pigment synthesized by melanocytes are eumelanin, or black/brown melanin, stored in eumelanogenic melanosomes, and pheomelanin, or yellow/red melanin, stored in pheomelanogenic melanosomes. Past study of the biogenesis of melanosomes was facilitated by the observation of four morphologically distinct stages of eumelanogenic melanosome development (Marks and Seabra, 2001; Seiji et al., 1963). Recent morphological studies were done which analyzed where melanosome biogenesis was blocked in melanocytes derived from HPS mice (Nguyen and Wei, 2004; Nguyen et al., 2002) and suggested an ordering for HPS protein function along the pathway of melanosome biogenesis (Figure 8).

HPS protein complexes function along the pathway of melanosome biogenesis. Mutations in genes encoding BLOC-1 components cause accumulation of stage I melanosomes and numerous aberrant vesicular structures lacking intralumenal striations, whereas defects in BLOC-2 components cause accumulation of stage I melanosomes and a novel melanosome intermediate, stage Ia, which is similar to stage I melanosomes in being vacuolar and not elliptical, but which contains intraluminal striations. BLOC-3 appears to function early in melanosome biogenesis as well, causing accumulation of stage I melanosomes. In mouse dorsal back follicular melanocytes, defects in BLOC-3 components introduce a mild rate limiting step and accumulation of stage I melanosomes (Nguyen et al., 2002), but in tail epidermal melanocytes, a more pronounced block in melanosome biogenesis is observed, so that mature melanosomes were relatively decreased in number (M.L. Wei and T. Nguyen, personal communication), suggesting that HPS proteins may differentially regulate melanocytes located in separate anatomic niches. Defects in the δ-subunit of AP-3 affect melanosome maturation between stage III and IV. Mutation of the Rabggta subunit of Rab geranylgeranyl transferase prevents the association of melanosomes with cortical actin filaments and impedes secretion of melanin particles into the extracellular space. BLOS3 is also known as HPS8/reduced pigmentation.
A model depicting trafficking pathways in the melanocyte, incorporating the data from HPS cells, together with recent studies on melanosome biogenesis and protein trafficking to the melanosome, is shown in Figure 9. At the trans-Golgi network (TGN), newly synthesized molecules destined ultimately for the lysosome or the melanosome are likely to undergo a first round of sorting. Molecules such as Pmel17 (a melanocyte-specific protein targeted to melanosomes and an integral component of the intra-lumenal fibrils) and MART-1/melan-a (a melanocyte-specific protein of unknown function) appear to be transported to the stage I melanosome, possibly via an AP1 mediated mechanism (De Maziere et al., 2002; Raposo et al., 2001). The syntaxin 13 molecule is localized to stage I melanosomes/coated endosomes (De Maziere et al., 2002; Prekeris et al., 1998), and the BLOC-1 subunit pallidin binds syntaxin 13 in yeast two-hybrid studies (Huang et al., 1999), suggesting a role for BLOC-1 at the level of the stage I melanosome, perhaps in mediating fusion events, as syntaxin 13 plays a role in endosomal fusion (Prekeris et al., 1998). Moreover, supporting a function for BLOC-1 at this early step in melanosome biogenesis are melanosome morphologies in the majority of BLOC-1 defective mice (pallid, cappuccino, muted, and sandy) that exhibited a block in melanosome maturation at the level of the stage I melanosome (Nguyen and Wei, 2004; Nguyen et al., 2002).

Model of trafficking pathways in melanocytes mediated by HPS proteins. Melanosomal proteins such as MART-1 and Pmel17 exit the TGN and traffic to the stage I melanosome/coated endosome via AP-1. Targeting and fusion of transport vesicles is possibly mediated by BLOC-1 binding to syntaxin 13. LAMP-1 and tyrosinase are transported to an early endosome via AP-1, then tyrosinase proceeds to later stage melanosomes via AP-3 and BLOC-3, whereas LAMP-1 continues on to late endosomes and lysosomes. BLOC-2 may mediate transport vesicle targeting, docking or fusion to later stage melanosomes and lysosomes. Tyrp1 is transported directly from the TGN to later stage melanosomes via BLOC-3 function. Alternatively, both tyrosinase and Tyrp1 could traffic to the late endosome via AP-3 and AP-1, respectively, and then traffic via BLOC-3 to the melanosome. After secretion of melanin, via fusion of the melanosomal limiting membrane with the plasma membrane, integral membrane molecules spanning the melanosome limiting membrane would remain on the cell surface, and are predicted to be internalized into endosomes and subsequently trafficked to lysosomes for degradation. Consistent with this are data which demonstrated MART-1, Pmel17 and Tyrp1 on the cell surface and also colocalizing with the lysosomal marker LAMP-1 (De Maziere et al., 2002; Levy et al., 2005; Raposo and Marks, 2002; Sprong et al., 2001). TGN, trans-Golgi network; MVB, multivesicular body.
Other molecules such as Tyrp1 and tyrosinase are likely targeted to later stage II or III melanosomes (Kushimoto et al., 2001; Raposo et al., 2001), but may arrive via distinct routes. Steady state distribution of tyrosinase was disrupted in AP-3-deficient melanocytes (Huizing et al., 2001b), but Tyrp1 was unaffected, suggesting that trafficking of tyrosinase is AP3-dependent and that of Tyrp1 is not. Tyrosinase appears to be targeted to melanosomes via an endosomal route, and seems to follow an initial pathway similar to that taken by the LAMP-1 and -3 molecules. In AP-3 deficient melanocytes, tyrosinase accumulated in multivesicular structures with tubular extensions resembling early endosomes (Huizing et al., 2001b) and in AP-3 deficient CTL, LAMP-3/CD63 also appeared to accumulate in endosomal structures, colocalizing with the early endosomal marker EEA1 (Clark et al., 2003). Studies on mouse fibroblasts deficient in AP-3 and AP-1 molecules have suggested that AP-1 mediates trafficking from Golgi to endosome and that AP-3 mediates trafficking from endosomes to lysosomes (Reusch et al., 2002). The data for melanocytes seems to be consistent with the findings in fibroblasts, with the extension that AP-3 mediates trafficking to melanosomes as well, as tyrosinase is mis-localized in the absence of AP-3 (Huizing et al., 2001b).
Genetic and morphological data in mice indicated that BLOC-1 mediated events likely precede BLOC-3 mediated events, and that BLOC-1 and BLOC-3 may work sequentially along the same pathway, as doubly homozygous pa/pa, ep/ep mice had the same pigmentary phenotype as BLOC-1 pallid mice (Nazarian et al., 2003). Immunoelectron microscopy studies co-localized Tyrp1 and AP-1 in the vicinity of the TGN and suggested that Tyrp1 may exit the TGN via an AP-1-mediated route and traffic directly to stage II melanosomes, persisting in stage III and IV melanosomes (Raposo et al., 2001). In cultured melanocytes derived from HPS-1 patients, defective in BLOC-3, Tyrp1 is found in large vesicles as well as in granular structures, compared with in control cells in which only the granular structures are noted (Richmond et al., 2005). Together, the data are consistent with Tyrp1 trafficking to early melanosomes from the TGN via an AP-1 and BLOC-3-dependent pathway. In the absence of HPS1, Tyrp1 may be arrested in the TGN or mislocalized to endosomal/lysosomal structures. The localization of HPS1 near the Golgi and on uncoated vesicles and on early melanosomes (Oh et al., 2000) suggests that HPS1 may play a role in the transport of vesicles destined for the melanosome. Ultrastructural analysis of melanosome morphology in mice defective in BLOC-3 is consistent with a role for BLOC-3 in melanosome biogenesis before the stage II melanosome, but after the delivery of Pmel17 to premelanosomes (Nguyen et al., 2002). In cells from HPS-1 patients, tyrosinase is also found in large vesicles, similarly to the effect on Tyrp1 (Richmond et al., 2005), again consistent with BLOC-3 having a role in vesicular transport to the melanosome.
In mice, the BLOC-2 subunit Hps6 is suggested to mediate fusion of transport vesicles to the plasma membrane (Oberhauser and Fernandez, 1996), and may likewise mediate fusion events at the level of the melanosome (and lysosome). In the absence of the BLOC-2 subunit HPS3, melanocytes from patients exhibited a more diffuse cytoplasmic distribution of tyrosinase, Tyrp1, LAMP-1, and LAMP-3, compared with a granular distribution in control cells (Richmond et al., 2005) and ultrastructural studies demonstrated an increased abundance of 50 nm vesicles containing pigment in the presence of dihydroxy-phenylalanine (DOPA), suggesting the intraluminal presence of either tyrosinase or Tyrp1 enzymatic activity (Boissy et al., 2005). The steady state distribution of Pmel17 and MART-1 were normal (Boissy et al., 2005). Thus BLOC-2 may mediate the fusion of transport vesicles containing tyrosinase or Tyrp1 with stage II or III melanosomes and vesicles containing LAMP-1 and LAMP-3 with melanosomes and/or lysosomes.
HPS effects on LROs
How have HPS defects affected other LROs, such as platelet dense bodies, lytic granules, and lung lamellar bodies? A common theme appears to be defects in secretion of many of these organelles. Skin cells from light ear mice (defective in HPS4) have decreased basal secretion of lysosomal hydrolases (Delprato et al., 2000). Platelets have abnormal thrombin stimulated secretion of dense granule contents (Novak et al., 1984). Lamellar bodies in lung alveolar type II epithelial cells in pale ear/pearl double homozygous mice (defective in HPS1 and AP-3 proteins) have decreased basal and ATP-stimulated secretory capacity for surfactant protein and phospholipids (Guttentag et al., 2005). A possible mechanism for the loss in secretory activity is suggested in HPS-1 and HPS-2, in which defective microtubule-meditated organelle motility is demonstrated for lysosomes in fibroblasts (Nazarian et al., 2003) and lytic granules in T cells (Clark et al., 2003), respectively. It could be that one or more motility factors, mediating attachment to or movement along microtubules, are missing or decreased in amount from the affected organelles in HPS, due to mis-targeting. Alternatively, selected HPS proteins may bind to these factors, which may be degraded in the absence of the HPS binding partner. Another possibility for lamellar bodies is that the process of exocytosis rather than motility is defective, and factors mediating events such as vesicle docking, targeting or fusion at the plasma membrane may be missing.
In the case of melanosomes, defects in Rabggta caused a marked block in secretion of melanin and an accumulation of intracellular melanosomes (Nguyen et al., 2002). However, in other HPS mouse strains an accumulation of melanosomes within melanocytes was not observed, and immature melanosomes were noted within neighboring keratinocytes, indicating that aberrant melanosomes were secreted to some extent (Nguyen and Wei, 2004).
In conclusion, the Hermanky–Pudlak syndrome is made up of a complex set of related autosomal recessive disorders caused by underlying defects in protein trafficking. Recent molecular, biochemical and cell biologic analyses together with clinical studies have provided insights into links between molecular and cellular pathology and clinical disease expression. Studies on HPS have led to a deeper understanding of basic cell processes, in particular mechanisms of cell-type specific specialized processes, led to the development of molecular tools to identify increasing numbers of HPS patients, and will likely help lead to targeted therapies for this currently untreatable and often fatal disease.
Acknowledgements
Many thanks to Drs Richard Swank and Edward Novak for critical reading of this manuscript and to Jerelyn Magnusson and Edgardo Caballero for excellent technical help.

