

Spatial genetic structure of aquatic bryophytes in a connected lake system
Editor: X.-Q. Wang
Abstract
Using genetic markers, we investigated the genetic structure of three clonal aquatic moss species, Calliergon megalophyllum Mikut., Fontinalis antipyretica Hedw. and F. hypnoides Hartm. on two scales: among populations in a connected lake system (large-scale spatial genetic structure) and among individuals within populations (fine-scale spatial genetic structure). Mean genetic diversities per population were 0.138, 0.247 and 0.271, respectively, and total diversities equalled 0.223, 0.385 and 0.421, respectively. Relative differentiation levels (FST values of 0.173, 0.280 and 0.142, respectively) were significant but showed that there is a moderate amount of gene flow taking place within the lake system connected with narrow streams. Bayesian STRUCTURE analysis provided some indication that the direction of water flow influences population genetic structuring in the studied aquatic mosses. We propose that dispersal leading to gene flow in C. megalophyllum, F. antipyretica and F. hypnoides takes place both along water via connecting streams and by animal vectors, such as waterfowl. Nevertheless, the slight genetic structuring pattern along the direction of water flow suggests that dispersal of shoots or their fragments along water is a means of dispersal in these mosses. The absence of sexual reproduction and spores may have caused the observed spatial genetic structure within populations, including aggregations of similar genotypes (clones or closely related genotypes) at short distances in populations otherwise showing an isolation by distance effect. Regardless of the results pointing to the dominance of vegetative propagation, it is impossible to completely rule out the potential role of rare long-distance spore dispersal from areas where the species are fertile.
Introduction
Two fundamental, intermingled events of plant ecology and evolution, dispersal and gene flow, are largely dependent on the reproductive mode, the efficiency of reproduction and on the mobility of the gametes and other propagules produced. Rates and patterns of dispersal and gene flow can be estimated either directly using ecological experiments or indirectly utilising molecular genetic markers. Even within a single plant species, dispersal and gene flow can vary considerably in space and time, depending on the environmental conditions (e.g. Pohjamo et al. 2006; Gonzales et al. 2010; Korpelainen et al. 2011). In addition, the role of dispersal vectors, such as wind, water and insects and other animals, may be considerable (Laaka-Lindberg et al. 2003; Tackenberg et al. 2003; Kowarik & Saumel 2008). In the case of restricted dispersal, gene flow is reduced, resulting in high spatial genetic structure (SGS), thus, formation of clusters of closely related individuals at a range of scales (Vekemans & Hardy 2004; Pohjamo et al. 2008; Korpelainen et al. 2011). In addition, SGS of plant populations may develop as a consequence of founder effects that occur at colonisation by a small number of individuals. However, if founder effects are the main cause of SGS, it should decrease or disappear with time (Ennos 2001).
Studies on bryophytes have shown that sexually produced spores are able to disperse further from the source than generally larger asexual propagules or shoot fragments (vegetative propagules), and that the size of the propagules matters (e.g. Sundberg 2005; Pohjamo et al. 2006; Korpelainen et al. 2011), while asexual propagules mainly contribute to the colony maintenance on a local scale (Laaka-Lindberg et al. 2003). However, it has been shown that asexual propagules may also contribute to long-distance dispersal to some extent (Pohjamo et al. 2006). As a consequence of the different dispersal patterns of sexually and asexually/vegetatively produced propagules, bryophytes with frequent sexual reproduction are genetically less differentiated due to a higher level of gene flow than mainly asexual ones (Korpelainen et al. 2005; Pohjamo et al. 2008).
Many species of bryophyte occupy aquatic or moist habitats, such as lakes, streams, bogs and springs. A fair number of bryophytes are found in association with freshwater, but there are no marine bryophytes and only a few species are found in brackish water (Longton 1992; Paton 1999; Ulvinen et al. 2002). Yet, most ecological and genetic studies on mosses and other bryophytes have concerned terrestrial species. Although aquatic bryophytes have attracted relatively little research interest among ecologists and geneticists (but see Hutsemekers et al. 2010), they are occasionally used as bioindicators (Vanderpoorten & Palm 1998; : Vanderpoorten 1999). Also, bryophyte communities typically segregate along an upstream–downstream gradient of habitat conditions and water quality, which promotes their use as direct bioindicators (Vanderpoorten 2003). For instance, Fontinalis antipyretica is used to monitor aquatic pollution (Vieira et al. 2009), to reveal the effects of UV-B radiation (Nunez-Oliveira et al. 2004) and to discover radionuclide contamination (Bolsunovsky 2004).
The aim of the present study was to investigate and compare the amount of genetic diversity and differentiation of three common aquatic moss species on two scales: (i) among populations within a connected lake system (large-scale spatial genetic structure), and (ii) among individuals within populations (fine-scale spatial genetic structure). The target moss species were Calliergon megalophyllum Mikut., F. antipyretica Hedw. and F. hypnoides Hartm. Of these, C. megalophyllum is monoecious and grows either submerged and attached to some substrate or float, but the two Fontinalis species are dioecious and grow attached to a substrate (Ulvinen et al. 2002). All three species are presumed to have rare spore production, and the main or only mode of reproduction in populations takes place through vegetative propagation. Therefore, as dispersal is expected to be reduced, we hypothesised that variation within populations is low, genetic distances between populations are high and relative differentiation among populations is high. We also expected that very narrow connecting streams between lakes (populations) and long distances create dispersal barriers, which would lead to geographic structuring of genetic variation among populations of C. megalophyllum, F. antipyretica and F. hypnoides.
Material and Methods
Taxon sampling and genotyping
Samples of C. megalophyllum, F. antipyretica and F. hypnoides were collected in Eastern Finland near the city of Kuopio (about 62º50′ N, 27º40′ E). The sampling area covered five small lakes and four connecting streams (Fig. 1). The downstream lake, Kivilampi, is connected to a large lake by a narrow stream, and the closest other lakes (unconnected to the studied lakes) are located at a distance of about 1 km from Lakes Vuorilampi and Kolmisoppi (information from the North Savo Regional Centre for Economic Development, Transport and the Environment). All found patches of the study species were sampled within the sampling area of five lakes and four streams, when the distance between samples of the same species was at least 10 m in order to avoid sampling the same genet twice. C. megalophyllum was found and sampled by diving or raking in four lakes, F. antipyretica in all five lakes and in three streams, and F. hypnoides in four lakes (Table 1). Because the occurrence (spatial coverage) of these three species varies widely within the study area, the sampling sizes were very uneven. No sporophytes were detected in the sampling area during the 3-day sampling period in late June and 2-day follow-up observations 1 year later.

The study area in Finland including five lakes and four connecting streams. The heights (m) of each lake above sea level are shown in parentheses, and the direction of water flow is marked with an arrow. Sampling details are given in Table 1.
| species | population | locality | sample size |
|---|---|---|---|
| Calliergon megalophyllum | CM-Ko | Kolmisoppi | 2 |
| CM-Ne | Neulalampi | 17 | |
| CM-Le | Levänen | 4 | |
| CM-Ki | Kivilampi | 6 | |
| Fontinalis antipyretica | FA-Vu | Vuorilampi | 2 |
| FA-VuKo | Stream between Vu-Ko | 3 | |
| FA-Ko | Kolmisoppi | 13 | |
| FA-KoNe | Stream between Ko-Ne | 1 | |
| FA-Ne | Neulalampi | 2 | |
| FA-Le | Levänen | 26 | |
| FA-LeKi | Stream between Le-Ki | 11 | |
| FA-Ki | Kivilampi | 7 | |
| Fontinalis hypnoides | FH-Ko | Kolmisoppi | 5 |
| FH-Ne | Neulalampi | 25 | |
| FH-Le | Levänen | 14 | |
| FH-Ki | Kivilampi | 3 |
Within a few weeks of collecting, DNA was extracted from dry plant material using the DNeasy Plant Mini Kit (Qiagen, Inc., Hilden, Germany) following the manufacturer’s instructions. DNA was then used for genetic fingerprinting with ISSRs (inter-simple sequence repeats) involving amplification by PCR, in separate reactions, with three arbitrary 17-base ISSR primers (tandem arrays of simple nucleotide motifs interrupted at one end by out-of-phase bases), which had been assembled by the Nucleic Acid – Protein Service Unit, University of British Columbia, Canada. Originally, 14 primers (primers 807–818 and 890–891 of set #9) were tested. The primers that resulted in most reproducible and variable band patterns were then selected for the study (5′–3′): 808, [AG]8C; 818, [CA]8G; and 891, HVH[TG]7. Amplification reactions were conducted in a volume of 20 μl. The reaction mixture contained about 10–20 ng genomic DNA, 1.2 units DyNAzyme II DNA polymerase (Finnzymes, Vantaa, Finland), 1× PCR buffer, 0.4 μl 10 mm dNTP mix and 1 μl of a single 5 μm primer. The thermocyler was programmed for 4 min denaturation at 94 °C, followed by 45 cycles of denaturation at 94 °C for 40 s, annealing at 50 °C (primer 891) or 52 °C (primers 808 and 818) for 40 s and elongation at 72 °C for 90 s. An additional 8-min elongation followed the last cycle. At the testing stage, amplification products were electrophoresed in 1.4% agarose gels and detected by staining with ethidium bromide. Final analyses for all samples were conducted using an automated analysis system of Agilent 2100 Bioanalyzer with DNA 7500 LabChip kits (Agilent Technologies, Santa Clara, CA, USA) using a fluorescent dye included in the kit. Reproducibility of banding patterns was confirmed by comparing duplicate reactions for about 50% of the samples.
Data analyses
In each species, the levels of genetic diversity (Nei 1987) in populations and total diversity were determined, and analysis of molecular variance (anova) with 1000 permutations was conducted based on the ISSR data (presence or absence of the polymorphic bands) using the ARLEQUIN 3.01 software (Excoffier et al. 2005; available at http://cmpg.unibe.ch/software/arlequin3/). As a result of amova, pair-wise FST values (Weir & Cockerham 1984) between samples and overall FST values were obtained. An estimate gene flow can be derived from Wright’s (1931) equation, as applied to haploids such as mosses, where FST is approximately 1/(1 + 2 Nm). Populations with sample sizes <5 were excluded from amova analyses.
Bayesian cluster analyses of population structure were conducted using the software STRUCTURE 2.2 (Pritchard et al. 2000; available at: http://pritch.bsd.uchicago.edu/structure.html) to determine the number of genetic clusters and the distribution of clusters in the populations. The software was used despite its assumption of populations being in the Hardy–Weinberg equilibrium (HWE) as such knowledge of HWE was not available for the studied haploid mosses. The used model was the correlated allele frequency model (Falush et al. 2003), which assumes that at each locus, allele frequencies are correlated. With this assumption, the model infers a population structure with K number of clusters based on the genotype data. An admixture model was applied. At each stage, analysis was repeated with different values of K (range 1–10) to discover the value with the highest estimate of log-likelihood probability (ln Pr(X|K) of the data. For each value of K, ten independent runs were conducted with burn-in length of 100,000 iterations, followed by a data collection period of 106 iterations. Furthermore, to identify the correct number of clusters (K) that best explain the data, ΔK values were calculated according to Evanno et al. (2005).
To discover the fine-scale spatial genetic structure (SGS) of populations, a spatial autocorrelation analysis was conducted as described in Vekemans & Hardy (2004) using the SPAGeDi 1.2 software (Hardy & Vekemans 2002; available at: http://ebe.ulb.ac.be/ebe/software.html) and calculating kinship coefficients (Hardy 2003). The multilocus kinship coefficient (F) values were regressed on the spatial distance between individuals using both spatial linear and natural logarithmic scales, providing regression slopes bd and bln. The significance of bd and bln against the null hypothesis H0: b = 0 (i.e. the absence of SGS) was tested by comparing the observed values with those obtained after 1000 random permutations of spatial positions of individuals. To discover whether SGS better matched predictions of isolation by distance in one dimension (kinship decreasing linearly with the linear distance) or predictions of isolation by distance in two dimensions (kinship decreasing linearly with the natural logarithm of the distance), the coefficients of determination, 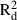 and
and 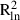 , were compared for the two types of regression (see, e.g. Born et al. 2008). To compare the pattern of SGS among samples on different spatial scales, mean multilocus kinship coefficients were computed for the whole dataset in each population, for all samples from each species and for the distance class 10–100 m.
, were compared for the two types of regression (see, e.g. Born et al. 2008). To compare the pattern of SGS among samples on different spatial scales, mean multilocus kinship coefficients were computed for the whole dataset in each population, for all samples from each species and for the distance class 10–100 m.
Results
Genetic composition and differentiation of populations
The ISSR primers 808, 818 and 891 used to analyse genetic variation and structure resulted in 66, 87 and 91 reproducible and polymorphic bands (loci) in the aquatic mosses C. megalophyllum (808: 22, 818: 24, 891: 20 bands), F. antipyretica (808: 32, 818: 27, 891: 28 bands) and F. hypnoides (808: 33, 818: 27, 891: 31 bands), respectively. The genetic diversity values were lowest in C. megalophyllum (total 0.223, average/population 0.138), while F. antipyretica and F. hypnoides contained very similar levels of variation (total 0.385 and 0.421, average/population 0.247 and 0.271, respectively; Table 2). The numbers of shared multilocus genotypes (MLG) varied: in F. hypnoides, seven out of 25 samples (28%) from FH-Ne (maximum pair-wise distance between shared MLGs 207 m) and seven out of 14 samples (50%) from FH-Le (maximum pair-wise distance between shared MLGs 396 m) shared the same MLG, while no shared MLGs were found in F. antipyretica or C. megalophyllum. The F. hypnoides populations with MLGs possessed considerable proportions of genetic diversity (0.361 and 0.382, respectively) when compared to the level found among all samples of the species (0.421).
| species | genetic diversity ± SD | FST value | |
|---|---|---|---|
| average/population | total | ||
| Calliergon megalophyllum | 0.138 ± 0.091 | 0.223 ± 0.179 | 0.173 |
| Fontinalis antipyretica | 0.247 ± 0.170 | 0.385 ± 0.141 | 0.280 |
| Fontinalis hypnoides | 0.271 ± 0.156 | 0.421 ± 0.107 | 0.142 |
The amova showed significant (P < 0.001) differentiation between all population pairs. The level of relative differentiation (FST) among populations equalled 0.173, 0.280 and 0.142 in C. megalophyllum, F. antipyretica and F. hypnoides, respectively (Table 2). The rest of the variation (0.827, 0.720 and 0.858, respectively) represented variation within populations.
The structure of genetic differentiation in each species including all populations was assessed using the Bayesian STRUCTURE analysis through determining the log-likelihood probability of the data (ln Pr(X|K)), as well as the rate of change values (ΔK). In C. megalophyllum, the highest, relatively even ΔK values were detected at K = 2 and K = 5 (Fig. 2A). Also in F. antipyretica, two distinct peaks were found at K = 2 and K = 4 (Fig. 2B). Thus, the analysis did not provide conclusive results for the number of clusters in these two species. However, in F. hypnoides, the best existing population structure was clearly captured with two clusters (K = 2) (Fig. 2C). The graphical presentation of inferred clusters in populations with sample size more than five show that in C. megalophyllum populations CM-Ne and CM-Ki are strongly differentiated: the cluster patterns at K = 2 are very similar but quite different at K = 5 (Fig. 2A), when CM-Ne has five sizeable clusters, but in CM-Ki two of those five clusters are mostly missing. The FST value between CM-Ne and CM-Ki was 0.173 (corresponding the gene flow estimate Nm=2.39), which also indicates considerable genetic differences. The cluster patterns of F. antipyretica are even more complex: populations FA-Ko and FA-Le (pair-wise FST = 0.104, corresponding the Nm estimate of 4.31) maintain fairly similar patterns also at K = 4, while populations FA-LeKi and FA-Ki (pair-wise FST = 0.388, corresponding the Nm estimate of 0.789) show similar patterns at K = 2, but very different ones at K = 4 (Fig. 2B). The clustering patterns of F. hypnoides with one ΔK peak at K = 2 indicates that populations FH-Ko and FH-Ne resemble each other, despite the high FST value 0.312 (corresponding the Nm estimate of 1.10), while population FH-Le shows a more distinct pattern (Fig. 2C); however, the sample size of FH-Ko was only five.

Inference of genetic clusters based on the software STRUCTURE. Mean (± SD) log-likelihood value of the data [L(K)] as a function of the K value (number of clusters), the mean change of the log-likelihood of the data (ΔK), and distribution of cluster assignment percentages shown in pie-charts for different K values in populations with sample size more than five based on ISSR data in A: C. megalophyllum, B: F. antipyretica and C: F. hypnoides.
Spatial genetic structure of populations
The mean and maximum pair-wise distances between samples in populations with enough samples (≥13) to allow an analysis of spatial genetic structure varied from 234 to 390 m, and from 572 to 799 m, respectively (Table 3). In analyses conducted for all samples from each species, the mean and maximum distances between samples ranged from 1069 to 1412 m, and from 2854 to 3848 m, respectively. Negative b values (i.e. regression slopes) were detected in all populations, although only the regression slopes of the two F. antipyretica populations were significant. All b values determined for the complete dataset were significantly negative (Table 3). Based on the coefficients of determination, the decrease of kinship fits the logarithmic distance slightly better than the linear distance, as expected for a SGS pattern in two-dimensional space. The 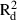 and
and 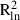 values varied from 0.005 to 0.108 and from 0.007 to 0.136, respectively.
values varied from 0.005 to 0.108 and from 0.007 to 0.136, respectively.
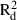 and
and 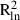 , respectively, based on all samples within each population.
, respectively, based on all samples within each population.| species | population | pair-wise distance (m) | bd (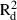 ) ) |
bln(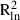 ) ) |
F | F10–100 | |
|---|---|---|---|---|---|---|---|
| mean | maximum | ||||||
| C. megalophyllum | CM-Ne | 312 | 781 | −0.005 (0.011) | −0.017 (0.007) | −0.012 | −0.008 |
| All | 1100 | 2913 | −0.003** (0.040) | −0.047** (0.055) | 0.000 | 0.084 | |
| F. antipyretica | FA-Ko | 360 | 754 | −0.007**(0.108) | −0.033**(0.136) | −0.009 | 0.078 |
| FA-Le | 390 | 799 | −0.006**(0.038) | −0.033**(0.052) | −0.010 | 0.051 | |
| All | 1412 | 3848 | −0.002*** (0.039) | −0.058*** (0.121) | 0.000 | 0.250 | |
| F. hypnoides | FH-Ne | 234 | 776 | −0.004 (0.005) | −0.018 (0.007) | −0.016 | 0.005 |
| FH-Le | 572 | 572 | −0.009 (0.027) | −0.031 (0.030) | −0.021 | 0.058 | |
| All | 2854 | 2854 | −0.066*** (0.044) | −0.016*** (0.056) | 0.000 | 0.201 | |
- F10–100 is the kinship coefficient for the distance class 0–100 m.
- Significance of b values is marked as *P < 0.05, **P < 0.01, ***P < 0.001.
Kinship coefficient (F) values based on all samples in each population were negative, ranging from −0.009 to −0.021, while kinship determined for all samples in each species equalled zero (Table 3). This shows that the kinship decreased with an increasing spatial distance within populations but that the genotypes had a more random distribution within the whole study area. However, the kinships determined separately for shorter distances (10–100 m) were positive, except in the C. megalophyllum population CM-Ne. This means that at relatively short distances, individuals of F. antipyretica and F. hypnoides populations are more closely related than expected.
Discussion
In Finland, there are 54 known aquatic moss species, which is about 8% of all moss species (Ulvinen et al. 2002). Thus, aquatic habitats harbour an important part of moss diversity. Because of the abundance of different aquatic habitats, most aquatic mosses, including C. megalophyllum, F. antipyretica an F. hypnoides investigated in this study, are abundant in Finland. Although there is a reasonable amount of knowledge of the means and rates of dispersal and gene flow in terrestrial bryophytes (e.g. Korpelainen et al. 2005, 2011; Sundberg 2005, 2010; Pohjamo et al. 2006, 2008), very little comparable information is available of aquatic species. The specific conditions of aquatic environments may contribute to the development of metapopulations with a high connectivity among subpopulations, as discovered in the present study on mosses, and also, for instance, in the seagrass Thalassia testudinum, which has been found to possess a metapopulation structure instead of spatially segregated and genetically distinguishable subpopulations (Bricker et al. 2011). On the other hand, the green alga Ulva prolifera is an example of an aquatic plant showing a very low level of gene flow among populations on a regional scale (Zhao et al. 2011).
Most likely, aquatic bryophytes mainly depend on water dispersal, and dispersal rates are influenced by water characteristics, primarily water flow. However, dispersal by animals may also occur. Birds and various invertebrates are known to act as transporters of algae (Atkinson 1972; Buschmann & Vergara 1993), and in the pondweed Potomogeton pectinatus population differentiation in the Baltic Sea has been found to be correlated with waterfowl dispersal (King et al. 2002). The role of birds as dispersal vectors of aquatic bryophytes was also proposed by Buch (1954), who suggested that some aquatic species, such as Ricciocarpus natans and Riccia fluitans, are dispersed in mud attached to the feet of waterfowl. McGregor (1961) actually found evidence that ducks disperse living bryophytes, as a fragment of Riccia fluitans was found attached to a feather at the back of the neck of a duck. In general, dispersal of bryophyte fragments is more likely to be successful, as live fragments can easily become caught on rocks or submerged roots and other material, while establishment through spores requires a suitable surface to allow germination and growth.
The present study provides tentative knowledge of the genetic diversity, differentiation and gene flow mechanisms in three aquatic moss species. As the occurrence of the species varied widely among the studied lakes and streams, the sample sizes varied considerably and the small sample sizes in some cases may have affected the outcome of the genetic analyses. Despite apparent clonal (vegetative) propagation, a reasonable amount of variation was found in individual populations of C. megalophyllum (mean genetic diversity 0.138), F. antipyretica (0.247) and F. hypnoides (0.271) when compared to total variation in each species (0.223, 0.385 and 0.421, respectively). Only two large clones possessing a shared multilocus genotype (MLG), both found in F. hypnoides. However, smaller clones were excluded on the grounds of the sampling strategy, as the distance between samples of the same species was at least 10 m. Yet, questions about genetic variation and clones are hampered by the lack of knowledge of the rates and origin of mutations in ISSR markers. The present variation may originate from sexual reproduction in a remote area or in a distant past, from rare sexual reproduction in the present populations or from more recent mutations. In previous studies on bryophytes, widely varying levels of genetic diversities have been discovered, e.g. a microsatellite variation range of 0.356–0.744 in six Sphagnum species (Shaw et al. 2008) but only 0.010 in a microsatellite study on S. wulfianum on a global scale (Kykjeeide et al. 2012), 0.059 in an ISSR study on Mannia fragrans on a regional scale (Hock et al. 2009), and ISSR diversity ranges of 0.035–0.168 in populations of Leptodon smithii (Spagnuolo et al. 2007a) and 0.098–0.175 in populations of Pleurochaete squarrosa (Spagnuolo et al. 2007b).
An Nm value of at least 1.0 is considered necessary to prevent divergence due to genetic drift (Wright 1931). In haploid organisms, like bryophytes, Nm ≥ 1.0 is obtained when FST ≤ 0.333. However, the mathematical model underlying the relationship between FST and Nm makes many assumptions, which may not be true in real populations (Whitlock & McCauley 1999). Thus, a FST value cannot be translated into an accurate estimate of Nm. Nevertheless, it provides an indicative estimate of the amount of gene flow. The FST values detected here in the monoecious C. megalophyllum (0.173), and in the dioecious F. antipyretica (0.280) and F. hypnoides (0.142) were all below the FST = 0.333 threshold, which means that despite apparent clonality and narrow connecting streams there is a fair amount of gene flow in the connected lake system, where this investigation was carried out. The FST values of C. megalophyllum, F. antipyretica and F. hypnoides were all in the range of differentiation values previously found in terrestrial mosses (reviewed by Korpelainen et al. 2005) Based on mostly allozyme, RAPD and microsatellite investigations in 30 terrestrial moss species, the average value of FST or comparable differentiation measures (GST, θST) equalled 0.234 (among commonly sexual and mainly clonal taxa 0.213 and 0.314, respectively), but there was large variation in the values (range 0–0.864) even in geographically local studies where the distances between populations are in the range of kilometres (Korpelainen et al. 2005). In a previous microsatellite marker-based study on the aquatic moss Platyhypnidium riparioides, which is presumed to disperse vegetatively, the detected differentiation among regional populations was high (FST = 0.57), and there was a correlation between genetic differences and geographical distances (Hutsemekers et al. 2010). It was suggested that one reason for such high differentiation might be that immigrants from some habitats are selected against in other habitats, reducing the effective migration rates. However, it is important to note that comparisons of FST values are truly meaningful only if similar markers are used, as otherwise differences in total genetic variation affect the values. In previous ISSR-based investigations on terrestrial bryophytes, varying differentiation levels have been detected, e.g. 0.046 and 0.067 in Pleurozium schreberi (Kotelko et al. 2008), 0.313 in Leptodon smithii (Spagnuolo et al. 2007a) and 0.411 in Pleurochaete squarrosa (Spagnuolo et al. 2007b), all on a regional scale.
Besides amova and other differentiation measurements, the Bayesian STRUCTURE analysis provided additional information of the population genetic structures. However, in C. megalophyllum and F. antipyretica the results were not conclusive. C. megalophyllum displayed two likely numbers of clusters (K) at 2 and 5. The species was sampled in all four sites of occurrence (populations) within the lake system consisting of five lakes and four connecting streams. The cluster numbers 2 and 5 developed through high genetic diversity and divergence detected in population CM-Ne, in which the genetic diversity equalled 0.228 (diversity across the species 0.223) and the mean genetic distance (proportion of average pair-wise differences) between individuals was 0.228. A part of population CM-Ne resembled population CM-Ko located upstream, while another part resembled populations CM-Le and CM-Ki downstream. Also F. antipyretica, which occurred in five lakes and three connecting streams, displayed two likely numbers of clusters, 2 and 4. As visible in Fig. 2B, there is an upstream cluster (shown for FA-Ko and FA-Le) and a downstream cluster (shown for FA-LeKi and FA-Ki). Yet, the overall differentiation is high (FST = 0.309) and the clustering pattern is complex. F. hypnoides, which occurred in four lakes, clearly showed the presence of two clusters, an upstream cluster (shown for FH-Ko and FH-Ne) and a downstream cluster (shown for FH-Le) (Fig. 2C). Thus, the direction of water flow is presumed to influence population genetic structuring in the studied aquatic mosses. However, as the studied lakes do not form a closed system, it is impossible to rule out the chance of dispersal, primarily by birds, from other lakes in the region. Also, the used STRUCTURE analysis assumes that the investigated populations are in Hardy–Weinberg equilibrium, although there is no knowledge of HWE in the case of the studied haploid mosses. Therefore, the obtained results should be considered with caution, despite previous examples of successful STRUCTURE analyses conducted even in populations showing HWE deviations (e.g. Schug et al. 2007),
We suggest that dispersal leading to gene flow in C. megalophyllum, F. antipyretica and F. hypnoides takes place both along water via connecting streams and by animal vectors, such as waterfowl. Yet, the slight genetic structuring pattern along the direction of water flow indicates that dispersal of shoots or their fragments along water is a means of dispersal in these mosses. Although monoecious plants have a tendency towards higher genetic differentiation (Hamrick & Godt 1997), that was not the case here. After all, the studied mosses rely completely or almost completely on vegetative propagation, and the role of the breeding system, dioecy or monoecy, is mostly irrelevant. C. megalophyllum, besides occurring attached to some substrate, may also float. However, there was no evidence of differing genetic structure or dispersal pattern due to the possibly easier movement of floating shoots. Regardless, it is impossible to completely rule out the potential role of rare long-distance spore dispersal from areas where the species are fertile.
The strength of spatial genetic structure (SGS) can be measured as the rate of decrease in genetic similarity with distance (Vekemans & Hardy 2004), and the SGS pattern provides indirect information on the amount of dispersal and gene flow. In the mosses C. megalophyllum, F. antipyretica and F. hypnoides, SGS varied clearly depending on the spatial scale: positive or close to zero kinship coefficients were found at short distances (up to 100 m), while the kinship coefficients determined for all samples in each examined population were negative and the kinships determined for complete datasets in each species equalled zero. Positive values indicate an aggregation of similar genotypes (clones or closely related genotypes), while negative values indicate the isolation by distance effect and individuals being less related than expected, presumably due to ineffective dispersal of vegetative shoots or their fragments. Kinships of zero indicate that genotypes have a more random distribution. No spore production is known to occur in the study area, and the detected SGS pattern indicates the absence of spores or their significant role in dispersal. Although two F. hypnoides populations (FH-Ne and FH-Le) each contained one widely distributed clone, range up to about 200 and 400 m, respectively, among other unique genotypes, this did not appear in the SGS pattern. Previously, Korpelainen et al. (2011) discovered in the liverwort Barbilophozia attenuata that the SGS pattern varied depending on the spatial scale and that SGS was strongly affected by the reproductive mode: sexual or asexual. In B. attenuata, asexual propagation by asexual gemmae possibly caused an aggregation of similar genotypes at short distances (<8 m), while at mid-distances (8–25 m) individuals were less related than expected, and at larger distances (>25 m) genotypes showed a more random distribution (kinship values about zero), apparently due to spores acting as effective means of dispersal. Thus, sexual reproduction and the production of spores or their absence may appear in the SGS pattern of bryophytes. When comparing the work on B. attenuata and the present study, there was a difference in the spatial scale: in the former study, the maximum pair-wise distance between samples was 81 m (also sporophyte production observed) while in the latter study maximum distances ranged between 2.9 and 3.8 km, depending on the species.
In this study, using genetic markers, we were able to reveal the genetic structure of clonal aquatic moss species, C. megalophyllum, F. antipyretica and F. hypnoides on two scales: among populations in a connected lake system (large-scale spatial genetic structure) and among individuals within populations (fine-scale spatial genetic structure). Contrary to our hypotheses, all species possessed a reasonable amount of genetic variation, both within populations and across populations, and differentiation levels showed that there is a moderate amount of gene flow taking place within the lake system. Thus the dominant role of vegetative propagation, the apparent absence of spore production and the lake morphology with narrow connecting streams did not lead to low dispersal, scarce genetic variation within populations and high differentiation among populations. However, the absence of sexual reproduction and spores may have caused the observed spatial genetic structure within populations, including aggregations of similar genotypes (clones or closely related genotypes; potentially novel mutations in ISSR marker regions) at short distances in populations otherwise showing isolation by distance effects.
Acknowledgements
We thank Hanna Forsman, Anna Kattan and Maria Pietiläinen for help in the laboratory, and we acknowledge financial support from Kuopion Luonnon Ystäväin Yhdistys ry.

