

Ectopic expression of phosphoenolpyruvate carboxylase in Vicia narbonensis seeds: effects of improved nutrient status on seed maturation and transcriptional regulatory networks
Summary
Seed maturation responds to endogenous and exogenous signals like nutrient status, energy and hormones. We recently showed that phosphoenolpyruvate carboxylase (PEPC) overexpression in Vicia narbonensis seeds alters seed metabolism and channels carbon into organic acids, resulting in greater seed storage capacity and increased protein content. Thus, these lines represent models with altered sink strength and improved nutrient status. Here we analyse seed developmental and metabolic parameters, and C/N partitioning in these seeds. Transgenic embryos take up more carbon and nitrogen. Changes in dry to FW ratio, seed fill duration and major seed components indicate altered seed development. Array-based gene expression analysis of embryos reveals upregulation of seed metabolism, especially during the transition phase and at late maturation, in terms of protein storage and processing, amino acid metabolism, primary metabolism and transport, energy and mitochondrial activity, transcriptional and translational activity, stress tolerance, photosynthesis, cell proliferation and elongation, signalling and hormone action and regulated protein degradation. Stimulated cell elongation is in accordance with upregulated signalling pathways related to gibberellic acid/brassinosteroids. We discuss that activated organic and amino acid production leads to a wide-range activation of nitrogen metabolism, including the machinery of storage protein synthesis, amino acid synthesis, protein processing and deposition, translational activity and the methylation cycle. We suggest that α-ketoglutarate (α-KG) and/or oxalacetate provide signals for coordinate upregulation of amino acid biosynthesis. Activation of stress tolerance genes indicates partial overlap between nutrient, stress and abscisic acid (ABA) signals, indicating a common interacting or regulatory mechanism between nutrients, stress and ABA. In conclusion, analysis of PEPC overexpressing seeds identified pathways responsive to metabolic and nutrient control on the transcriptional level and its underlying signalling mechanisms.
Introduction
Developing seeds differentiate from proliferating tissues into highly specialized storage organs. Maturation and storage activities are initiated during the transition phase, accompanied by fundamental transcriptional reprogramming and upregulation of genes related to storage metabolism (Radchuk et al., 2006; Ruuska et al., 2002; Sreenivasulu et al., 2004). Maturation is partly a response to exogenous and endogenous cues such as nutrient status, water and energy conditions, which are processed via a signalling framework involving SnRK1 kinases and abscisic acid (ABA), (Radchuk et al., 2006; Weber et al., 2005). SnRK1 in legume seeds works through ABA, which regulates a wide range of developmental events, mediates responses to stress and nutrients, and is necessary to proceed through seed maturation, to acquire desiccation tolerance and to prevent precocious germination (Finkelstein et al., 2002; Phillips et al., 1997). SnRK1-repressed pea seeds show maturation defects and have lower ABA contents (Radchuk et al., 2006; RJNE, RR, HW, unpubl. data). Macroarray-based gene expression analysis of pea SnRK1-antisense embryos revealed transcriptional upregulation related to mitotic activity, gibberellic acid (GA)/brassinosteroid (BR) synthesis, stress response and Ca2+-signal transduction. Downregulated genes are related to storage protein synthesis, stress tolerance, cell wall synthesis and ABA signal transduction. In summary, the phenotype resembles ABA-insensitivity/deficiency, and suggests that SnRK1 is a mediator of ABA function in pea seeds (Radchuk et al., 2006).
Understanding the regulation of seed maturation is particularly important for crops because of the initiation of storage metabolism. Legume seeds synthesize proteins and starch, and represent the most important source of protein for human and animal nutrition. Seed proteins are synthesized in the cotyledons during mid to late maturation. Amino acids and sucrose as precursors are unloaded from the phloem into the maternal seed tissues, from where they are transferred to the seed apoplast and subsequently taken up by the symplasmically isolated embryo. Storage protein synthesis is mainly controlled on the genetic level, and by nitrogen uptake and availability (Golombek et al., 1999; Miranda et al., 2001; Salon et al., 2001). Glutamine and/or asparagine are imported from the phloem. Uptake into soybean and pea embryos occurs in a partly passive way, especially during early stages; whereas a saturable system, via H+-amino acid co-transport, becomes important at later stages and is induced by N starvation, indicating control by assimilate availability (Bennett and Spanswick, 1983; Lanfermeijer et al., 1990). Much of the control of N uptake is embryonic. An amino acid transporter, VfAAP1, is expressed in Vicia faba cotyledon storage parenchyma cells, with maximum levels at the beginning of storage protein accumulation (Miranda et al., 2001). VfAAP1 overexpression in pea and Vicia seeds increased amino acid uptake and led to higher protein contents, which indicate that seed protein synthesis is N limited (Rolletschek et al., 2005).
Imported amides are frequently de-aminated in the legume seeds, and the ammonia-N is used to build other amino acids (Sodek et al., 1980). Thus, seed-specific amino acid biosynthesis requires carbon skeletons. The key acceptors are oxalacetate and α-KG, for the synthesis of the aspartate and glutamate family members, respectively. Any withdrawal of organic acids has to be compensated for by anaplerotic reactions. This requires coordination of the C/N metabolism. Phosphoenolpyruvate carboxylase (PEPC) fixes 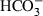 , and together with PEP yields oxalacetate, which can then be converted to other intermediates of the citric acid cycle, or can serve as a carbon acceptor for amino acid biosynthesis of the aspartate family. When pea embryos are pulse-labelled with 14CO2, the label appears almost entirely in malate, aspartate, citrate and glutamate, and later on in proteins (Flinn, 1985). Correlative evidence points to a rate-limiting role of PEPC in V. faba storage protein synthesis (Golombek et al., 2001). The highly regulated enzyme has allosteric properties, which are subject to opposite and antagonistic effects of metabolites like malate and Glc-6-P. Phosphorylation modulates the metabolic regulation with respect to feedback inhibition by malate (Golombek et al., 1999).
, and together with PEP yields oxalacetate, which can then be converted to other intermediates of the citric acid cycle, or can serve as a carbon acceptor for amino acid biosynthesis of the aspartate family. When pea embryos are pulse-labelled with 14CO2, the label appears almost entirely in malate, aspartate, citrate and glutamate, and later on in proteins (Flinn, 1985). Correlative evidence points to a rate-limiting role of PEPC in V. faba storage protein synthesis (Golombek et al., 2001). The highly regulated enzyme has allosteric properties, which are subject to opposite and antagonistic effects of metabolites like malate and Glc-6-P. Phosphorylation modulates the metabolic regulation with respect to feedback inhibition by malate (Golombek et al., 1999).
Transgenic Vicia seeds overexpressing Corynebacterium PEPC, which is not feed-back inhibited by malate, have larger seeds, increased seed protein content and show repartitioning of carbon from sucrose/starch to organic acids/free amino acids. These changes are consistent with an increased carbon flow through the anaplerotic pathway catalysed by PEPC, and indicate that seed storage protein accumulation is potentially limited by carbon acceptor availability (Rolletschek et al., 2004).
Our initial results (Rolletschek et al., 2004) show that PEPC overexpressing seeds have a greater storage capacity and increased seed protein content. The aim of this study is to analyse seed developmental and metabolic parameters of the PEPC-overexpressing line PPC-12, and the partitioning and distribution of both C and N over the whole plant. Line PPC-12 is one of the three lines independently generated, and displays the strongest phenotype. Nutrients and altered C/N status are putative signals for seed development. An array-based gene expression analysis was performed in order to identify respective metabolic pathways in the seed responding to metabolic control on the transcriptional level, and its underlying control and signalling mechanisms.
Results
Seed development and composition
Developmental parameters were compared for PPC-12 and wild-type embryos in terms of DW (Figure 1a) and FW accumulation (Figure 1b). The grey and black bars mark the transition and maturation stages. Physiological maturity, at which maximal seed DW is reached, occurred between 35 and 40 days after pollination (DAP) followed by desiccation. Both dry and FW accumulation display a similar pattern over time. Levels are not different from 15 to 22 DAP between PPC-12 and wild-type embryos. During early/mid maturation from 20 to 25 DAP, dry and FW accumulation is significantly lower in PPC-12 seeds, and is equal again at 30 DAP. Thereafter, at 35 DAP, accumulation rates are significantly larger in PPC-12 seeds compared with the wild type, where a plateau is approached. The FW to DW ratio reflects the water content of the embryo, and represents a measure for the developmental state. As DW accumulation proceeds there is a parallel decrease of the relative water content (Weber et al., 2000). For the PPC-12 embryos the ratio is higher during transition stage, between 15 and 17 DAP. However, the lag phase present in wild-type seeds is absent in PPC-12. From 20 DAP onwards PPC-12 embryos have always a higher FW to DW ratio (Figure 1c). Seeds from PPC-12 and the wild type are shown in Figure 1d.

Growth parameters of wild type and PPC-12 embryos.(a) DW, (b) FW and (c) ratio of FW to DW. Symbols are means (± SD), n = 4. Significant differences according to the Student’s t-test: *P < 0.05. DAP, days after pollination. Grey and black bars mark transition and maturation stages.
The data indicate a different development of PPC-12 seeds. Seed filling rates are lower at mid maturation but continuously higher at later stages, indicating that seed fill duration is longer but with lower rates. The higher FW to DW ratios of PPC-12 indicates delayed maturation.
Accumulation of sucrose and storage compounds was measured during maturation. Sucrose content in PPC-12 embryos was significantly higher at early maturation (19–21 DAP) and lower at mid maturation (25 DAP) (Figure 2a). There was a tendency for lower starch levels in PPC-12, but this was only significant at 19 and 25 DAP. Again, the lag phase in starch accumulation occurred only in the wild-type seed around 20 DAP (Figure 2b). Levels of extractable globulins (Figure 2c) and albumins (Figure 2d) were slightly higher for PPC-12, but this was significant only for globulins at 30 DAP.

Seed composition of PPC-12 embryos and the wild type during development.(a) Sucrose, (b) starch, (c) globulins and (d) albumins. Symbols are means (± SD), n = 4. Significant differences according to the Student’s t-test: *P < 0.05. DAP, days after pollination.
The data shows that especially at mid maturation (25 DAP) PPC-12 embryos contain less sucrose and starch, which was accompanied by increased globulins later on. This indicates a repartitioning of carbon from sucrose/starch to storage protein synthesis.
Nitrogen and carbon allocation
At mid maturation (28 DAP), dry matter, nitrogen and the partitioning of nitrogen were measured for the whole plant. Dry matter per plant, as well as seed dry matter per plant, was not different (Figure 3a,b). The percentage of nitrogen was higher in the vegetative parts of PPC-12 (Figure 3c), but at the same stage was not different for the seeds (Figure 3d). To measure uptake and allocation of nitrogen, PPC-12 and wild-type plants were pulse labelled with 6 mm of 15NH4 in 200 ml water that was applied to the roots. Four days later the plants were harvested and the 15N label was measured in vegetative organs and seeds. PPC-12 plants contained more label (Figure 3e) indicating higher uptake rates for nitrogen. The 15N content in the vegetative organs was higher for PPC-12 (Figure 3f). Moreover, the PPC-12 seeds revealed a more than twofold higher content of 15N, both in absolute terms per mg FW (Figure 3g) and on the per seed level (Figure 3h).

Partitioning of 15N tracer in PPC-12 and wild-type plants.(a) Dry matter per plant, (b) seed dry matter per plant, (c) percentage of nitrogen in vegetative organs, (d) percentage of nitrogen in seeds, (e) mg of 15 N tracer per plant, (f) mg of 15N tracer in vegetative organs, (g) mg of 15N tracer in seeds and (h) mg of 15N tracer per seed. Plants were labelled at mid-maturation (28 days after pollination). Bars are means (± SD), n = 5. Significant differences according to the Student’s t-test: *P < 0.05, **P < 0.01.
The results indicate higher nitrogen uptake into PPC-12 plants at mid maturation, which is distributed between vegetative organs and seeds. This is in accordance with altered seed sink strength in PPC-12.
In order to measure whole-plant carbon fixation and allocation between different organs, PPC-12 and wild-type plants at 28 DAP were in vivo pulse labelled with 13CO2 for 90 min following extraction after 4 days. In leaves, the recovered 13C label was not changed (Figure 4a). However, significantly more label was present in stems (Figure 4b) and in pods (Figure 4c), indicating that stems and pods may represent a temporary storage pool for carbon. Significantly more 13C label was also found in seeds (Figure 4d), indicating that more carbon has been transferred to PPC-12 seeds, and that the seed sink strength for carbon was higher. The data show that PPC-12 plants obviously fix more carbon, which is then partitioned into stems, pods and seeds.

Partitioning of 13C tracer in PPC-12 and wild-type plants.(a) Atom %13C tracer in leaves, (b) atom %13C tracer in stems, (c) atom %13C tracer in pods and (d) atom %13C tracer in seeds. Plants were labelled at mid-maturation (28 days after pollination). Bars are means (± SD), n = 5. Significant differences according to the Student’s t-test: **P < 0.01 and ***P < 0.001.
Seed acquisition of carbon and nitrogen
Variations in the distribution of stable isotopes of C and N can give evidence for isotopic discrimination of key enzymes, resulting in end products with different isotopic composition (Gleixner et al., 1998). Seed carbon mainly comes from two main CO2 fixing enzymes, Rubisco and/or PEPC. In contrast to Rubisco, PEPC does not discriminate between 13CO2 and 12CO2. Thus, a less negative δ13C value occurs when a higher percentage of carbon is fixed by PEPC (Melzer and O’Leary, 1987). The natural abundance of 15N is expressed as the δ15N value, and any differences reflect different nitrogen acquisition strategies or differences in nitrogen metabolism and losses. For example, plants relying only on N fixation have δ15N values close to zero, which is similar to the value for atmospheric N (Wanek and Arndt, 2002). To characterize the origins of C and N in PPC-12, a fraction of dry mature seeds was analyzed for total C and N, and for the abundance of N and stable C isotopes. As shown before, PPC-12 seeds contained more nitrogen (Figure 5a), as measured from dry matter, whereas the carbon content was unchanged (Figure 5b). The δ15N signatures were not different (Figure 5c). However, the δ13C value was less negative for the PPC-12 seeds (Figure 5d), indicating that these seeds contain more 13C. Therefore, the carbon in PPC-12 seeds may partly come from higher carbon fixation via PEPC in seeds.

Natural abundance of 15N and 13C in PPC-12 and wild-type mature seeds.(a) Percentage of total nitrogen in seeds, (b) percentage of total carbon in seeds, (c) δ13C in seeds and (d) δ15N in seeds. Bars are means (± SD), n = 5. Significant differences according to the Student’s t-test: *P < 0.05.
Differential gene expression in PPC-12 embryos
PEP carboxylase catalyses a side branch of the glycolytic pathway, and diverts carbon into the citric acid cycle. We showed that PEPC overexpressing seeds have higher individual seed weight, higher protein content and prolonged seed fill duration. Labelling experiments revealed that more carbon and nitrogen is partitioned into seeds. The PPC-12 seeds therefore represent models with improved nutrient status and increased seed sink strength.
We analysed differential gene expression in PPC-12 and wild-type embryos using macroarrays containing 5548 seed-specific genes from pea. Six developmental stages were analysed (at 15, 17, 19, 21, 25 and 30 DAP). Genes were regarded as differentially expressed when there were significant changes in either one (P < 0.05) or two (0.05 < P < 0.1) of the analysed stages. Using these criteria, a total of 192 genes (3.46%) with higher transcript abundance were identified and classified based on homology (Blast-2) and literature search, from which seven encoded histones (Figure S12) and 58 encoded ribosomal proteins (Table S1). We mention here that all following statements on gene identity and function must be considered as ‘putative’. Transcript abundances do not necessarily reflect transcriptional activity, protein content or enzyme activity. However, for simplicity, higher or lower transcript levels were referred to as up or down upregulated. Surprisingly, no genes were identified that met the criteria to be termed ‘downregulated’.
The 134 differentially upregulated transcripts, excluding ribosomal and histone genes (2.4%), were arranged into the following functional groups: storage proteins (9), endomembrane transport and protein processing (23), amino acid metabolism (17), metabolism and transport (12), energy and mitochondrial metabolism (5), transcriptional and translational activity (15), stress tolerance (14), photosynthesis (8), cell cycle and cell elongation (10), signalling and hormone action (13) and regulated protein degradation (18), see Table 1 and Figures S1–S12.
| ID number | Gene annotation | Species | Putative function |
|---|---|---|---|
| Storage proteins (9) | |||
| PSC27H17, PSC27M19, PSC25D19 | Legumin B | Faba bean | Storage protein |
| PSC21B12, PSC27A09 | Legumin A2 | Pea | Storage protein |
| PSC26M01, PSC29E16 | Legumin J | Pea | Storage protein |
| PSC24D15, PSC34G24 | Convicilin | Pea | Storage protein |
| Endomembrane transport & protein processing (23) | |||
| PSC28O02 | Syntaxin, NSF attachment protein (ALPHA-SNAP2) | Vitis | Vesicle trafficking |
| PSC27A02 | VPS29-like phosphoesterase protein | Arabidopsis | Vacuolar protein sorting |
| PSC33A22 | Peptidyl-prolyl cis/trans isomerase, PIN-1 | Arabidopsis | Protein folding, ER |
| PSS05C22 | SEC61 alpha | Arabidopsis | Protein translocation, ER |
| PSC31M06 | Signal sequence receptor, alpha subunit (TRAP) | Arabidopsis | Protein translocation, ER |
| PSS22N01, PSS05A01, PSC30C05 | Leucyl aminopeptidase | Petroselinum | Protein processing |
| PSC33N12 | Processing peptidase beta subunit | Tobacco | Protein processing |
| PSC29C04, PSC33B08 | Chaperone hsp70 | Medicago | Protein transport |
| PSS22M23 | Heat-shock protein 80 | Euphorbia | Molecular chaperone |
| PSC24G02 | Chaperonin CPN60-2 | Cucurbita | Molecular chaperone, Mitochondria |
| PSS07L05 | Chaperonin CPN10 | Arabidopsis | Molecular chaperone, Mitochondria |
| PSC28F07 | Chaperonin, δ-subunit | Soybean | Molecular chaperone, Cytosol |
| PSC30H20, PSC20L04, PSC24P14 | ADP-ribosylation factor | Arabidopsis | Vesicle trafficking |
| PSC28O19 | G-protein beta, sec13 | Arabidopsis | Protein transport |
| PSC30F03, PSC30J05, PSS13G04 | GTP-binding protein | Arabidopsis | Broad functions |
| PSC24B23 | Small G-protein ROP3 | Medicago | Vesicle trafficking |
| PSC34D06 | Small G-protein RAB5A | Lotus | Vesicle forming |
| PSS20N15, PSC28D03 | H+-ATPase proteolipid, vacuolar | Arabidopsis | Secretory functions |
| PSS05C12 | H+-Pyrophosphatase, vacuolar | Arabidopsis | Secretory functions |
| Amino acid metabolism (17) | |||
| PSC23O03 | Asparagine synthetase | Soybean | Asn synthesis |
| PSC29L12 | N-acetyl glutamate kinase 2 | Rice | Arg synthesis |
| PSC30J08 | Acetylornithine aminotransferase | Walnut | Arg synthesis |
| PSC27G19 | Argininosuccinate synthetase | Arabidopsis | Arg synthesis |
| PSS05L07 | Glutamate decarboxylase | Arabidopsis | GABA synthesis |
| PSC25H02 | Dihydrodipicolinate synth, DHDPS | Soybean | Lys synthesis |
| PSC33K22 | O-acetylserine-thiol-lyase | Rice | Cys synthesis |
| PSC35F03 | Serine acetyltransferase | Watermelon | Cys synthesis |
| PSC25P16 | Acetohydroxy acid isomeroreductase | Pea | Ile, Val synthesis |
| PSS15E11 | Methionine synthase | Medicago | Met synthesis |
| PSC31E22 | S-adenosylmethionine synthetase | Medicago | |
| PSS15E01, PSC25O14, PSS09O11 | S-adenosyl-l-homocysteine hydrolase (AdoHcyase) | Medicago | Methylation cycle |
| PSC24D16 | S-adenosylmethionine decarboxylase β | Medicago | Methylation cycle |
| PSC25O17 | DNA (cytosine-5-)-methyltransferase | Pea | DNA methylation |
| PSC28A16 | Formate-tetrahydrofolate ligase | Medicago | C1 transfer |
| Transcriptional & translational activity (15) | Translation | ||
| PSC27P05 | Elongation factor 2 (EF-2) | Sugar beet | Translation |
| PSC29O18 | Elongation factor 1 | Rice | |
| PSC29J04, PSS09E23, PSS18B10 | Translation initiation factor eIF3 γ subunit | Arabidopsis | Translation |
| PSC25B22 | Translation initiation factor eIF2 γ subunit | Arabidopsis | Translation |
| PSS07I14 | Translation initiation factor 1A (EIF-1A) | Arabidopsis | Translation |
| PSC25I24 | Nascent polypeptide associated complex (NAC-α) | Rice | Translation |
| PSC30J06 | Splicing factor | Chickpea | Splicing |
| PSC28F02, PSS18F04, PSC23E08 | DEAD/DEAH box RNA helicase | Arabidopsis | Transcription |
| PSC27C04 | Small nucleolar RNPs, GAR1 | Arabidopsis | Translation |
| PSS20B07, PSC31M05 | PolyA binding protein | Tobacco | Transcription/translation |
| Primary & secondary metabolism & transport (12) | |||
| PSC26G07 | Phosphofructokinase, PP-dep. | Rizinus | Glycolysis |
| PSC33L09, PSC31N21 | GAPDH, cytosolic | Pea | Glycolysis |
| PSS12L01 | Pyruvate kinase, cytosolic | Arabidopsis | Glycolysis |
| PSS15P22 | NADP-ICDH, cytosolic | Medicago | CH metabolism |
| PSC27O09 | Beta-ketoacyl-ACP synthetase I-2 | Soybean | Fatty acid synthesis |
| PSC25O09 | Adenylate kinase | Arabidopsis | Nucleic acid synthesis |
| PSS23O04 | Dihydroorotate dehydrogenase | Rice | Nucleic acid synthesis |
| PSC28H03 | Ribonucleoside-P2 reductase small chain | Tobacco | Nucleic acid synthesis |
| PSS07L07 | GLc-6-P translocator | Arabidopsis | Plastidial transport |
| PSC23C16 | Outer plastidial membrane protein, PSPOR1 | Pea | Plastidial transport |
| PSS06I10 | Sucrose synthase 1 | Pea | Sucrose cleavage |
| Energy & mitochondrial & metabolism (5) | |||
| PSS09L14 | Mitochondrial F1-ATPase, γ | Arabidopsis | ATP synthesis |
| PSS11J04, PSS24E18 | NADH-ubiquinone oxidoreductase subunit | Arabidopsis | Respiration |
| PSC35G10 | Ubiquinol-cytochrome C reductase, subunit 1 | Tobacco | Respiration |
| PSC33M24 | Frataxin | Arabidopsis | Regulation of mitochondrial functions |
| Stress tolerance (14) | |||
| PSC31D16 | Superoxide dismutase Cu/Zn | Pea | ROS scavanging, Cytosol |
| PSC28M14 | Superoxide dismutase | Pea | ROS scavanging, Mitochondria |
| PSC33F04 | 2-cys peroxiredoxin | Pea | Redox homoestasis |
| PSS20L12 | Cationic peroxidase | Chickpea | Redox signalling |
| PSS06C21 | Leu-rich repeat resistance protein | Millet | Resistance |
| PSC23A09 | Mismatch binding protein Mus3 | Maize | DNA repair |
| PSC25B09 | Pyridoxine biosynthetic enzyme | Bean | Broad functions in stress tolerance |
| PSC31K03 | NIMA-related protein kinase | Populus | Broad functions in stress tolerance |
| PSC22H11, PSS07H15, PSC24I02, PSC30E08, PSS09N16 | Alcohol dehydrogenase 1 | Pea | Broad functions in stress tolerance |
| PSC26L10 | UDPG:flavonol 3-O-glycosltransferase | Tobacco | Broad functions in stress tolerance |
| Photosynthesis (8) | |||
| PSS24C19, PSC28B22 | PS I, psaD | Spinach | PS light reaction |
| PSC32H14 | PS I, psaH | Tobacco | PS light reaction |
| PSC27N02 | PS I psaF, reaction center subunit III | Vigna radiata | PS light reaction |
| PSS07G10 | PS II, psbO, O2-evolving complex 33 kDa | Spinach | PS light reaction |
| PSC24N22 | Chlorophyll A–B binding protein (LHCII type I) | Pea | PS light reaction |
| PSC35E12 | Rubisco subunit binding-protein β | Arabidopsis | Calvin cycle |
| PSS17A23 | Transaldolase | Tomato | Calvin cycle |
| Cell cycle & cell elongation (10) | |||
| PSC24C23 | Actin | Soybean | Cytokinesis |
| PSS05L01, PSS13M16 | Tubulin | Arabidopsis | Cytokinesis |
| PSC33G10 | Prohibitin | Arabidopsis | Role in cell division |
| PSS22I04 | Profilin | Bean | Actin binding |
| PSS24G17 | Histone deacetylase | Soybean | Chromatin modification |
| PSC22J12, | Auxin-induced protein SAUR | Arabidopsis | Regulation of IAA/BR responses |
| PSC22E19 | Auxin-induced protein GH1 | Soybean | Regulation of IAA/BR responses |
| PSC26L24 | Aquaporin | Medicago | Cell expansion |
| PSC34A05, PSC33A15 | Hydroxyproline-rich glycoprotein 1 | Pea | Cell wall synthesis |
| Signalling & hormone action (13) | |||
| PSC30D03 | Transcription factor Abi-3 | Pea | Transcriptional regulation |
| PSC34F22 | Transcription factor (PHD zinc finger) | Arabidopsis | Transcriptional regulation |
| PSC34L09 | Transcription factor | Faba bean | Transcriptional regulation |
| PSC28L12 | Transcription factor | Faba bean | Transcriptional regulation |
| PSC23A03 | Transcription factor (bHLH) | Arabidopsis | Transcriptional regulation |
| PSC32P08 | Transcription factor (HD-Zip) | Arabidopsis | Transcriptional regulation |
| PSC27A23 | Transcription factor 3 RNA polymerase B | Arabidopsis | Transcriptional regulation |
| PSC30N14 | Transcription factor AP2/EREBP | Arabidopsis | Transcriptional regulation |
| PSC34C03 | Cytochr. P450 monooxygenase, CYP714A1 | Arabidopsis | Hormone synthesis |
| PSC22K07 | BR biosynthetic protein LKB | Pea | BL synthesis |
| PSS07I03 | Adenosine kinase | Tobacco | Cytokinin synthesis |
| PSC03H08 | Phospholipase C phosphoinosite-specific | Pea | Signal transduction |
| PSC33A12 | 14-3-3 protein | Tomato | Broad role in enzyme regulation |
| Regulated protein degradation (8) | |||
| PSC35K02, PSC30D12, PSS10O14, PSC23E03 | 20S proteasome subunit | Arabidopsis | Regulated protein degradation |
| PSC25B20 | 26S proteasome reg su | Arabidopsis | Regulated protein degradation |
| PSS07N10 | Ubiquitin extension protein | Potato | Regulated protein degradation |
| PSC29N24, PSC03H20 | Ubiquitin-conjugating enzyme | Arabidopsis | Regulated protein degradation |
| PSC22G21 | Ubiquitin | Arabidopsis | Regulated protein degradation |
| Histones (7) | |||
| PSS23D21, PSS21A22, PSC34G07 | Histone H2A | Pea | DNA binding |
| PSS06O05, PSC32E09 PSC32F09, PSS07B13 | Histone H4 | Wheat | DNA binding |
| Ribosomal protein genes (58) see Table S1 | |||
- ER, endoplasmic reticulum; BR, brassinosteroid; ATP, adenosine 5′-triphosphate; ROS, reactive oxygen species; indole-3-acetic acid (IAA).
Upregulated genes involved in protein storage activity and amino acid biosynthesis
In general, gene expression in the wild type occurs in a biphasic manner separated by a lag phase from 18 to 20 DAP. In PPC-12 seeds, this lag phase has frequently disappeared. A group of nine storage protein genes was upregulated: seven legumins and two convicilins. The individual profiles are shown in Figure S1. The summarized profiles reveal that gene expression is similar for PPC-12 and wild-type embryos at 15–19 DAP, and higher in PPC-12 seeds at 18–20 DAP and again after 25 DAP (Figure 6a).

Expression profiles of selected upregulated genes in the PPC-12 embryos (red colour) and in the wild type (blue colour). Axes: x-axis, days after pollination; y-axis, relative expression units (log of normalized signal intensities).(a) Legumin B (PSC27H17, PSC27 M19, PSC25D19), legumin A2 (PSC21B12, PSC27A09, legumin J (PSC26 M01, PSC29E16) and convicilin (PSC24D15).(b) Chaperone hsp70 (PSC29C04, PSC33B08), heat-shock protein 80 (PSS22 M23), chaperonin CPN60-2 (PSC24 G02), chaperonin CPN10 (PSS07 L05), chaperonin and δ subunit (PSC28 F07).(c) ADP-ribosylation factor, (PSC30 J05, PSC30H20), signal sequence receptor (PSC31 M06) and H+-ATPase (PSS20 N15).(d) Elongation factors (EF-2) (PSC27P05, PSC29O18), translation initiation factors (PSC29 J04, PSC25B22, PSS09E23), nascent polypeptide associated complex α (PSC25I24), RNA binding protein A (PSS09B02), splicing factor (PSC30 J06), DEAD/DEAH box RNA helicase (PSC28 F02), GAR1 protein (PSC27C04) and poly(A)-binding protein (PSS20B07).(e) N-acetyl glutamate kinase 2 (PSC29 L12), acetylornithine aminotransferase (PSC30 J08) and argininosuccinate synthetase (PSC27 G19).(f) S-adenosyl-l-methionine synthase (PSC31E22) and S-ado-homocysteine hydrolase (PSC25O14, PSS09O11, PSS15E01).(g) Ribonucleotide reductase R2 (PSC28H03) and dihydroorotate dehydrogenase (PSS23O04).(h) Alcohol dehydrogenase 1 (PSC22H11, PSS07H15, PSC24I02, PSC30E08, PSS09 N16).(i) Photosystem 1 (PS 1); psaD (PSS24C19); PS 1, psaH (PSC32H14); PS 1, psaF, reaction center subunit III (PSC27 N02); PS 1, psaD PS 1 chain II (PSC28B22); PS 2, psbO O2-evolving complex 33 kDa (PSS07 G10); chlorophyll A–B binding protein 8 precursor (LHCII type-1 CAB-8) (PSC24 N22); Rubisco subunit binding-protein β subunit (PSC35E12); transaldolase (PSS17A23).(j) Actin (PSS07I13) and tubulins (PSS13 M16, PSS05 L01).(k) Transcription factors ABI-3 (PSC30D03) and transcription factor HD-ZIP (PSC32P08).(l) Transcription factors AP2/EREBP (PSC30 N14), PHD zinc finger (PSC34 F22), VfPTF1 (PSC28 L12).
A group of 23 upregulated genes are involved in endomembrane transport, vesicle biogenesis and protein processing (Figure S2, Figure 6b,c). Syntaxin play key roles in cellular processes of vesicle trafficking, fusion and secretion. Vacuolar protein sorting (Vps29) are components of the retromer complex required for efficient sorting of seed storage proteins (Shimada et al., 2006). Three genes may be involved in protein translocation and processing in the endoplasmic reticulum (ER): SEC61, forming ER translocation channels; signal sequence receptor protein (TRAP), regulating the retention of ER resident proteins; Pin1-type peptidyl-prolyl cis/trans isomerase, which is involved in ER protein folding. Four other genes encode protein-processing peptidases. Six genes represent different chaperones and chaperonines involved in various aspects of protein processing and translocation across membranes (Figure 6b). Three sequences encode ADP-ribosylation factors (Arfs), which are major regulators of vesicle biogenesis in intracellular trafficking (Memon, 2004). Six genes encode G proteins that have a wide role in vesicle trafficking (Molendijk et al., 2004). Another three genes encode vacuolar isoforms of H+-ATPase (adenosine 5′-triphosphatase) proteolipid and H+-pyrophosphatase, which energize vacuolar transport processes (Maeshima, 2001). In summary, the results indicate that different aspects of storage protein synthesis, maturation, deposition and vesicle trafficking are stimulated upon PEPC overexpression. This is in accordance with the upregulation of 15 genes involved in transcriptional and translational activity (Figures S3 and 6d): two encode elongation factors, five represent subunits of translation initiation factors and eight other proteins are involved in RNA splicing, transcription, processing and translation. The profiles of gene expression show upregulation between 17 and 21 DAP (Figure 6d), and are similar for genes involved in endomembrane transport (Figure 6b).
A total of 17 genes encode enzymes involved in amino acid metabolism. Three are involved in serine/cysteine biosynthesis: plastidic O-acetylserine-thiol-lyase, serine acetyltransferase and formate-tetrahydrofolate ligase (Figure S4). The latter catalyses the folate-mediated C1 transfer via ATP-dependent activation to 10-formyl tetrachydrofolate (THF), which is important for the synthesis of methionine and purines, and for glycine/serine interconversion (Hanson and Roje, 2001). Another four enzymes are associated with glutamate metabolism. Three genes, involved in the biosynthesis of arginine, are upregulated: N-acetyl glutamate kinase 2, acetylornithine aminotransferase and argininosuccinate synthetase (Figure 6e). Arginine is the most frequent amino acid in pea seed proteins (Slocum, 2005). Four enzymes are upregulated within the aspartate pathway: asparagine synthetase, dihydrodipicolinate synthase, acetohydroxy acid isomeroreductase and methionine synthase. Remarkably, eight upregulated sequences encode enzymes involved in the plant methylation cycle (Figures 6f), which is responsible for the production and the utilization of methyl groups (Gallardo et al., 2002; Radchuk et al., 2005).
To analyse whether upregulated expression of amino acid biosynthesis genes influenced the levels of free amino acids, we analysed five stages of embryo development of both PPC-12 and wild type. As shown in Figure 7 the sum of free amino acids in PPC-12 embryos was clearly increased by around twofold between 17 and 22 DAP, whereas no clear difference occurred between 26 and 30 DAP. The higher levels are mainly caused by increases of the most abundant amino acids: arginine, alanine, asparagine, glutamate and glutamine. Some others are not largely altered, like aspartate, leucine and lysine. These results together indicate a wide-range activation of amino acid and storage protein biosynthesis.

Levels of free amino acids in PPC-12 embryos and wild-type embryos during maturation. Data points are means from four replications ± SD.
Other functions upregulated in PPC-12 embryos
A group of 12 genes upregulated in PPC-12 embryos encode enzymes of the primary/secondary metabolism and of assimilate transport (Figure S5). Five genes encode enzymes of sucrose breakdown and glycolysis: sucrose synthase 1, PPi-phosphofructokinase, pyruvate kinase, and two glycetaldehyde-3-phosphate dehydrogenase (GAPDHs). Their upregulation indicates increased glycolytic carbon flux in the PPC-12 seeds caused by increased PEPC activity. Three genes are involved in nucleotide metabolism: dihydroorotate dehydrogenase, ribonucleoside-diphosphate reductase (Figure 6g) and adenylate kinase. β-Ketoacyl-ACP synthetase is involved in lipid synthesis. Two upregulated genes encode plastidial assimilate transporters: Glc-6-P-translocator and PsPOR1, an outer plastidial membrane protein. NADP-iso-citrate dehydrogenase (NADP-ICDH) is a key cytosolic enzyme that links C and N metabolism (Hodges et al., 2003).
A group of five genes are involved in energy and mitochondrial metabolism (Figure S6). Four are involved in the mitochondrial electron transport chain and ATP synthesis (complex 1, 3 and 5). Frataxin is required for maintenance of mitochondrial homeostasis (Busi et al., 2004). Increased organic acid production by PEPC overexpression is a possible reason for the upregulation of mitochondrial metabolism.
Another 14 genes are involved in stress response and/or stress tolerance (Figure S7). Five encode alcohol dehydrogenase 1 (ADH1) (Figure 6h), and which in Arabidopsis are ABA-inducible and related to drought tolerance (De Bruxelles et al., 1996). Another two sequences are related to DNA repair and genotoxic stress tolerance: Mus3 mismatch repair protein and NIMA-related (non-inherited maternal antigen) protein kinase (Noguchi et al., 2002). Another gene UDP-glucose:flavonol 3-O-glucosyltransferase is homologous to bronze-1 in maize, a key enzyme of anthocyanin biosynthesis, which has protective functions. Four genes are involved in reactive oxygen species (ROS) scavenging, and are potentially stress and ABA responsive, and are induced during seed maturation and desiccation (Aalen, 1999; Bailly, 2004). Two other genes are involved in broad functions of stress and disease tolerance: leucine-rich repeat resistance protein and pyridoxine biosynthetic enzyme.
A group of eight upregulated genes are related to photosynthesis (Figure S8). Six are involved in photosynthesis light reactions: four subunits of photosystem 1 (PS 1), one of PS 2 and one Chl A/B binding protein. Another sequence represents transaldolase. In the PPC-12 embryos the expression profiles of photosynthesis-related genes reveal generally higher levels at early development, and between 19 and 21 DAP and at 30 DAP (Figure 6i).
Ten upregulated genes are related to cell cycle and cell elongation (Figure S9). The profiles show upregulation in PPC-12 embryos between 15 and 19 DAP (Figure 6j). There are two tubulins and one actin, which are associated with the microtubular system, and are frequently present during cell division and elongation of Medicago embryos (Gallardo et al., 2003). One gene encodes profilin, an actin binding protein, and another encodes prohibitin, which is involved in cell-cycle control, signalling and mitochondrial homeostasis (Chen et al., 2005). Upregulated histone acetylase can modify chromatin structure and is required for progression through mitosis in tobacco (Li et al., 2005). An auxin-binding protein (GH1) can regulate IAA responses. Aquaporins are necessary for solute uptake for cell expansion. Two isoforms of hydroxyproline-rich glycoproteins are upregulated. The expression of these cell-wall proteins is critical for cell shape, the correct positioning of cell plates during cytokinesis and normal embryo development (Hall and Cannon, 2002).
Upregulated signalling and hormone functions
A group of 13 genes is upregulated with roles in signalling and hormone action (Figure S10). Eight encode transcription factors. Two of them (ABI-3 and HD-ZIP; Figure 6k) reveal a similar mode of upregulation and expression profile as the storage protein genes, indicating some control on storage protein gene expression (Figure 6a,k). ABI-3, a well-known key regulator of seed maturation, confers ABA sensitivity and activates storage protein gene promoters (Wobus and Weber, 1999) and cellular differentiation in response to ABA and sugars (Rohde et al., 2002). Another upregulated sequence encodes phospholipase C, which in Arabidopsis is increased under stress and plays a role in secondary ABA responses (Liu et al., 2006). Three others, AP2/EREBP, PHD zinc finger and VfPTF1 are upregulated during early stages (Figure 6l), and their profiles more closely resemble the genes involved in cell elongation (Figure 6j) or arginine biosynthesis (Figure 6e). Three other transcription factors that are also upregulated may also play a role in early seed development. One encodes a basic helix-loop-helix (bHLH) transcription factor, which regulates cell elongation and may modulate GA signalling (Kim et al., 2005). Another upregulated gene is related to BR action and encodes BR biosynthesis protein LKB from pea, a homologue to DIMINUTO/DWARF-1, which converts 24-methylenecholesterol to campesterol (Nomura et al., 1999), and is together with GA involved in cell elongation. Another sequence encodes cytochrome P450 monooxygenase (CYP714A1), which is possibly involved in GA catabolism (Zhu et al., 2006). The results indicate that signalling pathways related to GA/BR and ABA are upregulated in PPC-12 embryos. One gene encodes adenosine kinase (ADK), which is important to remove adenosine in order to maintain S-adenosyl-methionine (SAM) utilization and recycling, and also plays a role in cytokinin synthesis (Moffatt et al., 2002). Adenosine levels were measured in PPC-12 and wild-type embryos. Levels were significantly lower in PPC-12 at 15 DAP, indicating increased degradation by ADK, which is upregulated on transcript level between 15 and 19 DAP. However, adenosine levels were not different at later stages (Figure 8a, Figure S5). Another sequence encodes 14-3-3 protein, which can coordinate primary carbon and nitrogen metabolism (Comparot et al., 2003).

Levels of abscisic acid (ABA) in PPC-12 and wild-type embryos.(a) Levels of adenosine in PPC-12 and wild-type embryos. Bars are means (± SE), n = 3. (b) Levels of ABA. Bars are means (± SE), n = 4. Significant differences according to the Student’s t-test: *P < 0.05, **P < 0.01.
Another eight genes are related to regulated proteolysis via the ubiquitin system (Figure S11), which may be involved in phase transitions during seed development. Four genes encode subunits of the 20S proteasome, and one encodes a 26S proteasome subunit. Another two encode ubiquitin-conjugating enzyme and one encodes ubiquitin extension protein.
Finally, seven genes encoding histones 2A, B and 4 (Figure S12) and 58 ribosomal protein genes are also found to be upregulated (Table S1).
To validate the results obtained by macroarray hybridization, a subset of seven of the upregulated genes were analysed in growing embryos by northern gel hybridization using the same developmental stages. Figure S13a shows the results for N-acetyl glutamate kinase (PSC29 L12), acetylornithine aminotransferase (psc30 J08), photosystem II, psbO (PSS07 G10), argininosuccinate synthase (PSC27 G19), syntaxin (PSC28O02), adenosine homocysteine hydrolase (PSS09O11) and legumin B (PSC27H17). Northern blot results were quantified and displayed together with the array results (Figure S13b). The results demonstrate accordance between array and Northern blot hybridization results.
ABA levels in wild-type and PPC-12 embryos are not different
In seeds ABA is necessary to proceed through maturation and to acquire stress tolerance. It has been shown that nutrient responses are linked to the responses of ABA (Wobus and Weber, 1999). Several mutants affected in ABA synthesis and sensitivity are sugar-sensing mutants, indicating that sugar or nutrient signalling requires an intact ABA transduction chain (Rook et al., 2001).
Activation of storage protein genes and, tightly connected, of stress tolerance in the PPC-12 seeds could be explained by increased ABA levels in the embryos. We therefore measured the levels of ABA in growing wild-type and PPC-12 embryos of the same stages as used for the macroarray hybridization. Levels were significantly higher in PPC-12 embryos at 15 DAP before storage activity starts, and slightly lower at 17 DAP. However, during maturation there were no large differences (Figure 8b), indicating a metabolic effect on ABA sensitivity rather than on its synthesis.
Discussion
Phosphoenolpyruvate carboxylase overexpressing seeds have a greater storage capacity and an increased seed protein content. Thus, the transgenic seeds represent suitable models to study metabolic control of seed metabolism and composition, and altered seed sink strength. In this study we analysed seed developmental parameters of PPC-12 seeds, and the partitioning and distribution of both C and N for the whole plant. Measuring the changes of global gene expression in response to improved nutrient status and sink strength revealed metabolic and developmental pathways responding to metabolic control on the transcriptional level, as well as putative signalling mechanisms.
PPC-12 embryos take up more carbon and nitrogen
To analyse differences in seed acquisition for carbon and nitrogen, δ15 N and δ13C signatures were determined. The δ13C values were less negative meaning that the content of 13C is higher in PPC-12 seeds. Assuming that the seed carbon is finally derived from the two main CO2 fixing enzymes, PEPC and Rubisco, and that only Rubisco discriminates between 13C and 12C, the less negative δ13C values coincide with a higher percentage of C fixation by PEPC in the PPC-12 embryos.
PPC-12 seeds contain more nitrogen. However, the δ15N values are not different, which argues against a larger participation of N nodule fixation for N acquisition. This is not surprising because the plants were grown under non-limiting N conditions, which may suppress nodule N fixation. However, the PPC-12 seeds have higher total N contents, which indicates increased N uptake. This is in accordance with our finding that after 15N ammonium pulse labelling to the roots at mid seed filling, more 15N is taken up and partitioned to vegetative organs and seeds (Figure 3f,g). We can speculate further that the higher demand of nitrogen of the PPC-12 seeds is largely covered by stimulation of root uptake, probably via some whole-plant signalling of nitrogen demand (Gansel et al., 2001). Studies with Arabidopsis (Lejay et al., 1999) have also shown that the nitrogen status of the whole plant controls uptake of N via long-distance signalling. Collectively, the labelling studies show that PPC-12 embryos take up more C and N, and can thus be considered as models with altered sink strength.
PEPC overexpression alters seed development
Seed filling rates of PPC-12 are lower at early to mid maturation, but continuously higher at later stages. This is in accordance with the higher seed water and sucrose content, indicating a physiologically younger stage. Thus, seed fill duration is longer but rates are lower at mid maturation.
Collectively, these results and those from the gene expression studies (Figure 6) reveal that PPC-12 seed development differs mainly during two periods, 17–21DAP and 25–30 DAP. In the wild type a lag phase is present at 17–21 DAP, which is evident at different levels, in the FW to DW ratio (Figure 1c), during starch accumulation (Figure 2b) and in the gene expression profiles (Figures 6 and S13). Such lag phases frequently occur in seed development of different species, and are probably connected to developmental phase transitions and/or genetic reprogramming (Sreenivasulu et al., 2004; Weber et al., 2005; Weschke et al., 2000). Generally, in legumes and others seeds, like barley, the lag phase separates cell division/elongation from maturation/storage product accumulation. It is tempting to speculate that the disappearance of the lag phase in the PPC-12 seeds, which in particular is evident on the transcriptional level (Figure 6), could be to the result of increased sink strength caused by PEPC overexpression.
At late maturation (26 DAP), lower sucrose levels in PPC-12 embryos can result from the increased consumption of carbohydrates required for organic acid and amino acid formation. The levels of both are higher in PPC-12 embryos (Rolletschek et al., 2004; Figure 7). This is also in accordance with the higher globulin levels found at 30 DAP (Figure 2d) and with the N content in mature seeds (Rolletschek et al., 2004).
PEPC overexpression stimulates cell elongation
The individual seed weight of PPC-12 seeds is larger, which, however, is not reflected on the total seed mass per plant, which is unchanged. Increased individual seed weight may be explained by a changed course of cell elongation and dry matter accumulation. Activation of cell elongation is evident on the transcriptional level between 15 and 22 DAP (Figure 6j). From the genes involved, several are related to the microtubular system. Besides these structural genes (one actin, two tubulins, profilin and two aquaporins), stimulation of cell elongation is also consistent with upregulated genes related to GA/BR biosynthesis and function: CYP714A1, BR biosynthetic protein LKB and several GTP binding proteins. A bHLH transcription factor (PSC23A03) may regulate cell elongation and modulate GA signalling (Kim et al., 2005). The transcription factors AP2/EREBP (PSC30 N14), PHD zinc finger (PSC34 F22) and VfPTF1 (PSC28 L12) showing a similar pattern of upregulation (Figure 6l,j) could be involved in cell elongation. In Arabidopsis a member of AP2/EREBP has been shown to affect seed size (Ohto et al., 2005), and another member is involved in regulation of Arabidopsis seed metabolism (Cernac and Benning, 2004).
Taken together, the results indicate that growth stimulation of PPC-12 seeds occurs mainly via cell expansion. This would then lead to the observed larger seeds and to a higher storage capacity. In contrast, storage protein gene expression (Figure 6a) and globulin accumulation (Figure 2a) are not different at 15–17 DAP, and starch accumulation is even lower (Figure 2b). The possible stimulation of cell elongation relative to storage product synthesis can also explain the larger seed fill duration, evident by the extended DW accumulation (Figure 1a) and the higher transcript levels of storage protein genes after 25 DAP (Figure 6a).
Our results reveal stimulated expression of 192 genes in the PPC-12 embryos. Such changes could have at least two different origins: changed seed development or overexpression of PEPC.
PEPC overexpression leads to the wide-range activation of storage protein synthesis
We showed that PEPC overexpressing seeds channel more carbon into organic acids, which would then be available for amino acid and storage protein biosynthesis. Gene expression analysis reveals a wide-range activation of the metabolism of different nitrogen compounds in PPC-12 embryos, which includes the machinery of storage protein synthesis, amino acid synthesis, protein processing and deposition, translational activity and the methylation cycle. The upregulation of a larger number of ribosomal genes also points to stimulated protein synthesis.
Nine storage protein genes are upregulated, from which seven are legumins. This is in accordance with increased globulin levels observed earlier (Rolletschek et al., 2004). Similarly, pea seeds overexpressing amino acid permease have increased globulin content (Rolletschek et al., 2005), and gene expression analysis reveals that from 17 upregulated storage protein genes, eight encode legumins and, interestingly, again no vicilin gene is activated (K. Weigelt et al., unpubl. data). However, in the SnRK1-repressed pea seeds, displaying an ABA-insensitive/deficient phenotype, vicilins (six isoforms) and vicilin-like unknown seed protein (USP) (four isoforms) are particularly affected (Radchuk et al., 2006). Therefore, it is tempting to speculate that in Vicia/pea seeds vicilin gene expression is more strongly controlled via an ABA-dependent pathway, whereas legumin gene expression is more under nutritional control.
During seed maturation, the precursors of storage proteins are synthesized on the rough ER and are sorted to protein storage vacuoles by vesicle-mediated machinery, where they are converted into the mature forms (Muntz, 1998; Robinson et al., 2005). Storage protein deposition, as well as cell division and expansion, all require the synthesis and trafficking of membranes, proteins and polysaccharides through the network of organelles. A total of 23 upregulated genes are involved in endomembrane transport, vesicle biogenesis and protein processing. However, secretory membrane trafficking can also be involved in a variety of other plant-specific processes, including ABA and auxin signalling, plant development, tropic responses and pathogen defence (Carter et al., 2004). Comparing the expression profiles of the storage protein genes (Figure 6a) with those of endomembrane transport (Figure 6c) and of chaperones (Figure 6b) reveals different clusters, and indicates that these genes can also be related to other processes, for example cell elongation (Figure 6j). The profile of others, such as syntaxin, GTP binding protein (PSC30 G03) and G protein Rop3, are more similar to that of the storage protein genes, pointing to a role of these candidates in storage protein deposition.
Stimulation of organic acid production leads to the wide-range activation of N metabolism
The gene expression analysis revealed a massive induction of amino acid metabolism at the transcriptional level. This is in good accordance with increased levels of individual amino acids during PPC-12 embryo maturation. Also in Arabidopsis a coordinated upregulation was shown for metabolites and transcripts (Fait et al., 2006). Ten genes encode enzymes directly involved in amino acid biosynthesis (schematic overview in Figure 9). Two enzymes, serine acetyltransferase and O-acetylserine-thiol-lyase participate in the serine branch and can produce cysteine from serine. A putative sink for cysteine is also the methylation cycle, which is upregulated on the transcriptional level (Figure 10). Formate-tetrahydrofolate ligase is a C1 donor required for serine glycine interconversion, but has also roles in other reactions like purine biosynthesis and the methylation cycle. Four enzymes are each involved in the aspartate and glutamate branch of amino acid metabolism (Figure 6). The carbon skeleton of aspartate is oxalacetate, which is the direct product of PEPC. Glutamate derives from α-KG, which can be produced inside the TCA cycle from oxalacetate or directly by cytosolic NADP-ICDH, which is also upregulated in the PPC-12 embryos. It has been suggested that cytosolic NADP-ICDH is important in supplying C skeletons for N assimilation and/or amino acid biosynthesis by equilibrating and maintaining cellular levels of isocitrate and α-KG. Its transcripts are nitrate and/or sucrose inducible (Hodges et al., 2003). Our results confirm the important role for NADP-ICDH in seeds supplying carbon acceptors for amino acid biosynthesis.

General scheme of pathways of amino acid biosynthesis in seeds, after Macnicol (1977). Overexpression of phosphoenolpyruvate carboxylase (PEPC) is given in light red. Amino acids of the cysteine, aspartate and glutamate branch are highlighted in orange, green and blue, respectively. Enzymes that are upregulated on the gene expression level are given in red. For details see the text.

General scheme of methylation cycle in plants. Enzymes that are upregulated on the gene expression level are given in red. For details see the text.
Three enzymes in the glutamate branch are involved in the synthesis of arginine, N-acetylglutamate kinase (NAGK), N-acetylornithine amino transferase and argininosuccinate synthetase (Figure 9). Arginine biosynthesis in seeds is of particular importance. This amino acid represents up to 40% of the total N in seed storage proteins and is the most abundant free amino acid in pea seeds, and is catabolized as an N source only during germination. The entire arginine pathway is plastid localized (Slocum, 2005). In pea cotyledons NAGK is allosterically regulated by arginine (McKay and Shargool, 1981). A novel mechanism suggests that arginine synthesis is modulated in response to changes in the global C/N status, with NAGK being the key enzyme (Forchhammer, 2004). In a range of plants NAGK is bound and activated by the plastid-localized PII protein, which has been shown to be a key mediator of cellular responses to C and N (Ninfa and Jiang, 2005). The role of the PII protein is to sense the cellular C, N and energy status, and to confer this information to other proteins through protein-protein interaction. The primary function of PII in Arabidopsis is to relieve feedback inhibition by arginine after binding ATP and α-KG (Chen et al., 2006). Arabidopsis knock-out mutants of PII show reduced ornithine, citrulline and arginine accumulation (Ferrario-Mery et al., 2006). Therefore, in the PPC-12 seeds, it is possible that α-KG, which production requires part of the glycolysis, PEPC, partial TCA cycle and ICDH, represents a signal of the C/N status, and that a PII protein serves as a sensor, which then activates arginine biosynthesis. Because PEPC overexpression in seeds causes stimulation of organic acid production (Rolletschek et al., 2005), it is tempting to speculate that in the PPC-12 seeds α-KG and/or oxalacetate act as the primary signal for a coordinated upregulation of amino acid biosynthesis. Such a metabolic signal coordinating the C/N metabolism is well established for bacteria (Galvez et al., 1999; Ninfa and Jiang, 2005), whereas evidence in seeds is scarce. At least for Arabidopsis plants there is evidence that the PII system links and coordinates carbon and nitrogen metabolism (Chen et al., 2006; Ferrario-Mery et al., 2006).
We analysed gene expression of a PII homologue, cloned from pea seeds, and found strong upregulation in wild-type cotyledons at the onset of maturation (T.M. Meitzel, R.S. Radchuk and H.W. Weber, unpubl. data). However, the PII function in seeds with respect to regulation of C/N interactions must be defined in more detail in the future.
PEPC overexpression stimulates the methylation cycle
Eight genes are upregulated that participate in the methylation cycle, which is fed by the amino acids methionine, cysteine and aspartate (Thomas and Surdin-Kerjan, 1997). Its function is to supply SAM for transmethylation reactions and the synthesis of ethylene and polyamines (Gallardo et al., 2002; Radchuk et al., 2005). For a general scheme see Figure 10. Methionine synthase, S-adenosyl homocysteine hydrolase (AdoHyc), S-adenosyl methionine synthetase and ADK are all upregulated at early phases, indicating a need for SAM at that stage. Upregulation of a methyltransferase at later stages can be important for DNA methylation during endopolyploidization, which occurs in parallel with cell elongation (Radchuk et al., 2005). Methyltransferase reactions are efficiently inhibited by S-adenosyl-homocysteine (SAH), which has to be steadily removed by AdoHcy from which three isoforms are upregulated. AdoHyc itself is feedback-regulated by adenosine, which can be degraded by ADK which is also upregulated.
In the PPC-12 seeds SAM decarboxylase and Met2 DNA methyltransferase are upregulated at late stages. In plants SAM decarboxylase activity can determine the levels of the polyamines spermidine and spermine (Thu-Hang et al., 2002). These polyamines are known to be linked to stress situations, and it is generally accepted that they have protective characteristics. Stimulation of polyamine biosynthesis at later stages of seed development is in accordance with the acquisition of stress tolerance during maturation. However, there is no evidence for interactions of polyamine synthesis with the ‘stress hormone’ ABA. In stressed tomato leaves polyamines and ABA act independently (Kim et al., 2005).
Activation of stress tolerance indicates partial overlap between nutrient, stress and ABA signals
A total of 14 genes related to stress tolerance are upregulated, from which five encode ADHs (Figure 6h). Seed maturation is generally known to be associated with the acquisition of stress tolerance, which goes in parallel with storage product deposition. Both functions are positively regulated by ABA. The profiles of Abi-3 (Figure 6k), the storage proteins (Figure 6a) and the ADHs (Figure 6h) are similar. Recent studies with SnRK1-repressed seeds, which are ABA deficient (Radchuk et al., 2006; RJNE, RR and HW, unpubl. data) reveal that a subset of genes behaves reciprocally compared with PPC-12 seeds. In the SnRK1-repressed seeds, eight genes related to stress tolerance are downregulated (Radchuk et al., 2006). In the PPC-12 seeds six of them are found to be upregulated. Also, transcription factors Abi-3 and HD Zip are upregulated in PPC-12 embryos (Figure 6k), and downregulated in SnRK1-repressed embryos. Because the primary effect in the PPC-12 seeds are improved sink strength and nutrient status, a common interacting or regulatory mechanism may exist between nutrient, stress and ABA action. The increased ABA levels at 15 DAP in the PPC-12 embryos are unclear at the moment, but may be derived from maternal organs, as shown for Arabidopsis (Frey et al., 2004). However, during maturation ABA concentrations are not increased, which speaks against a direct influence of the nutrient status on ABA synthesis in the PPC-12 seeds. Another explanation can be a metabolic effect on ABA sensitivity. It has been suggested that nutrients increase ABA sensitivity or levels. During Arabidopsis germination exogenous sugars can modulate internal ABA concentration, either increasing its synthesis or inhibiting its degradation (Price et al., 2003). Alternatively, ABA could enhance the ability of the cell to respond to sugars/nutrients (Rook et al., 2001). However, this knowledge mainly comes from seedling analysis, whereas for seeds little information is available.
Experimental procedures
Plant material
The transgenic line PPC-12, Vicia narbonensis (Rolletschek et al., 2004), was grown in 2-L pots in growth chambers under a light/dark regime of 16-h light (20°C) and 8-h dark at (18°C). Plants were fertilized once a week with nitrate and ammonium in order to keep non-limiting nitrogen conditions. For the isolation of embryos, pods were tagged according to the number of DAP, collected in the middle of the light phase and processed further. For biochemical analysis seeds were harvested, and embryos were immediately isolated and snap-frozen in liquid nitrogen.
Extraction and determination of sucrose, starch, protein, and total C and N
After ethanol extraction sucrose levels were determined enzymatically. The starch-containing insoluble material was solubilized in 1 N KOH for 1 h at 95°C and neutralized with 5 N HCl. Starch was hydrolyzed with amyloglucosidase and determined enzymatically. To determine albumin and globulin fractions of extractable proteins, powdered samples were extracted in acetate buffer [50 mm acetate, 1 mm KCl, 10% (v/v) DMSO, 0.5% (v/v) butanol; pH 4.5] and, subsequently, in phosphate buffer (100 mm KH2PO4, 100 mm Na2HPO4, 500 mm KCl; pH 7). Proteins were measured with bovine serum albumin as standard. Relative contents of total carbon and nitrogen in dried, powdered samples of cotyledons were measured using an elemental analyzer (Vario EL; Elementar Analysensysteme, http://www.elementar.de). Statistical analysis was performed using a Student’s t-test using sigma stat software (Jandel Scientific, Erkrath, Germany, http://www.systat.de).
Extraction and determination of free amino acids
Freeze-dried plant samples were pulverized, extracted twice in 80% ethanol at 60°C and centrifuged. Samples were dried and redissolved in 20 mm HCl and derivatized using the AccQ-Tag method (Waters Associates, http://www.waters.com) and run on a reversed-phase HPLC system (Waters Associates). The HPLC system consisted of a gradient pump (600), a degassing module, an autosampler (717) and a fluorescence detector (474). Chromatograms were recorded using the software millennium 2010. Gradient was accomplished with buffer A containing 140 mm sodium acetate (pH 5.8; Suprapur; Merck, http://www.merck.com) and 7 mm triethanolamine. Acetonitrile and water were used as eluents B and C. Amino acids were separated on reversed-phase column AccQ Tag, 150 × 3.9 mm, equilibrated with buffer A at a flow rate of 1 ml min–1 and heated at 37°C during the whole measurement. The gradient was produced by the following concentration changes: 1 min, 1% B, 0.4% C; 27 min, 5% B, 10.7% C; 28.5 min, 9% B, 11.3% C, 35 min, 12.5% B, 13.8% C; 45 min, 24.5% B, 17.8% C; 47.5 min, 60% B, 40%C; 50.5 min, 0% B, 0%C; 60 min, 0% B, 0% C.
In vivo labelling with 13C
In order to measure whole-plant carbon fixation and allocation between different organs, PPC-12 and wild-type plants at 28 DAP were covered with transparent plastic bags (40 × 150 cm) and pulse labelled with 13C barium carbonate, 99 atom%13C for 60 min. 13CCO2 was generated by injecting 5 ml of 2 m HClO4 into a beaker with 1 g Ba13CO3. Plastic bags were removed and plants were cultivated for 4 days before extraction and measurement of 13C in the different organs. The 13C measurements were performed by K. D. Wutzke, Research Laboratory, Children’s Hospital, University of Rostock, Germany using the mass spectrometer Tracer mass 20-20 (SerCon, http://www.sercongroup.com), and total 13C as well as the 13C concentration was calculated.
15N-labelling experiments
A 200-mL volume of 6 mm15NH4Cl (Chemotrade GmbH, Leipzig, Germany, http://www.chemotrade-leipzig.de) was applied to the roots of AAP-12 plants at 28 DAP. Sampling of all seeds was performed 4 days later. Samples were dried for 2 days at 60°C and ground so as to pass through a 0.5-mm sieve. For the determination of atom %15N, the remaining solution of NH4Cl following titration (Kjeldahl N analysis) was evaporated. The remaining solution was adjusted to an N concentration of app. 500 μg ml–1, and the enrichment of 15N was determined by emission spectrometry (Isonitromat 5200; Statron, http://www.statron.de), see also Götz and Herzog (2000). All values of 15N and 13C are expressed on an excess basis, which means tracer minus background isotope.
13C and 15N signatures
Aliquots of the dry plant material (2 mg) were weighed into tin capsules and analyzed for total N, C and atom %15N and 13C by continuous-flow gas isotope ratio mass spectrometry (CF-irM). The CF-irMS system consisted of an elemental analyser coupled to a gas isotope ratio mass spectrometer (DeltaPLUS; Finnigan, MAT, http://www.thermofinniganmat.de).
Determination of ABA and adenosine
ABA and adenosine were extracted, purified and quantified by an isotope dilution assay using the extraction methods of Ferguson et al. (2005). 2H4 ABA (NRC-PBI, http://pbi-ibp.nrc-cnrc.gc.ca) and 13C5 adenosine (Omicron Biochemicals Inc., http://www.omicronbio.com) were added as quantitative internal standards. Purification included mixed-mode reverse-phase cation exchange (Oasis MCX-SPE column; Waters), pre-conditioned with 5 ml of CH3OH followed by 5 ml of 1.0 m HCOOH. The sample was loaded in 1.0 m HCOOH and ABA was eluted with 5 ml of CH3OH. Adenosine was eluted with 5 ml 0.35 m NH4OH in 60% (v/v) CH3OH. Purified ABA and adenosine were analyzed by [LC-(+)ESI-MS/MS] using a Waters 2680 Alliance HPLC system (Waters) linked to a Quattro-LC triple quadrupole MS (Micromass, Altrincham, UK; see Ferguson et al., 2005). Compounds were quantified by multiple reaction monitoring (MRM) of the mother (parent) ion and daughter (product) ions, as described in Ross et al. (2004) for ABA. The conditions for adenosine were as follows: cone voltage, 26 V; collision energy, 30 eV; MRM channels were 273 > 136 for 13C5 adenosine, and 268 > 153 for adenosine.
cDNA macroarray data evaluation and Northern gel blot analysis
A description of the macroarray is given in Radchuk et al. (2006). Hybridization experiments were performed twice with independent biological replicates. The ArrayVision signal was the basis for all subsequent analyses. Intensities of individual spots and of corresponding local backgrounds were determined. Data analysis was normalized and filtered as described by Radchuk et al. (2006), with additional modification at filtering steps. Limit value maximum/minimum ratio for double spots for intensities higher than three times the background was set to 1.5. Limit value for SD for each of the normalized spot intensities was set to 0.5. The limit value was increased by 0.15 and 0.30 for lower intensities (two and one times background). To assess the significance of differentially expressed genes in PPC-12 embryos, a screening was performed for twofold and higher relative expression ratios, followed by a paired sample one-sided t-test with risk levels P < 0.05 in at least one stage analysed, or 0.05 < P < 0.1 for at least two stages analysed. The complete data set was reduced to sequences showing significant expression. The EST annotation was performed with BLASTx2 with E values < 1E−20. The macroarray results were confirmed by northern analysis of selected genes (Figure S13).
Visualization and analysis of the gene expression data set was performed using the VANTED platform (Klukas et al., 2006). After the completing the rounds of filtering steps, the non-logarithmic data set were put onto the graph elements. The raw dataset of the two experiments and all EST sequence data, along with additional information, is available at Array Express (http://www.ebi.ac.uk/arrayexpress; login, E-IPKG-2; password, 1163422229894).
RNA isolation and gel blot analysis was performed as described by Heim et al. (1993).
Acknowledgements
We are grateful to Katrin Blaschek, Elsa Fessel, Angela Schwarz and Susanne Moryson for excellent technical assistance. We thank Isolde Saalbach for the help in plant transformation and Ulrich Wobus for discussions and continuous support. We thank Uwe Scholz, Thomas Rutkowski and Christian Klukas for support in Bioinformatics, and Tobias Meitzel for the help collecting plant material. We acknowledge the help of Ursula Tiemann and Karin Lipfert for figure artwork. This work was supported by the European Union (Integrated project GRAIN LEGUMES), the Deutsche Forschungsgemeinschaft (WE 1641/9-1) and NSERC, Canada.

