

QTL detection on porcine chromosome 12 for fatty-acid composition and association analyses of the fatty acid synthase, gastric inhibitory polypeptide and acetyl-coenzyme A carboxylase alpha genes
Summary
Refinement of previous QTL on porcine chromosome 12 for fatty-acid composition and a candidate gene association analysis were conducted using an Iberian × Landrace cross. The concentrations of ten fatty acids were assayed in backfat tissue from which four metabolic ratios were calculated for 403 F2 animals. Linkage analysis identified two significant QTL. The first QTL was associated with the average chain length ratio and the percentages of myristic, palmitic and gadoleic acids. The second QTL was associated with percentages of palmitoleic, stearic and vaccenic acids. Based upon its position on SSC12, fatty acid synthase was tested as a candidate gene for the first QTL and no significant effects were found. Similarly, gastric inhibitory polypeptide (GIP) and acetyl-coenzyme A carboxylase alpha (ACACA) were tested as candidate genes for the second QTL using three SNPs in GIP and 15 synonymous SNPs in ACACA cDNA sequences. Two missense SNPs in GIP showed significant effects with palmitoleic and stearic fatty-acid concentration. Highly significant associations were found for two SNPs in ACACA with stearic, palmitoleic and vaccenic fatty-acid concentrations. These associations could be due to linkage disequilibrium with the causal mutations.
Introduction
Dietary fatty acids are highly relevant nutrients for human health including saturated fatty acids (SFA) and mono- and poly-unsaturated fatty acids (MUFA and PUFA) respectively. SFA with 14 or 16 carbons increase plasma cholesterol; however, MUFA and PUFA have a hypocholesterolemic effect and increased intake is associated with reduced risk of cardiovascular diseases (Lichtenstein 2006). Fat from meat is primarily composed of MUFA and SFA, in which oleic acid [C18:1(n-9)] is the most abundant fat source, followed by palmitic (C16:0) and stearic (C18:0) acids (Valsta et al. 2005).
Fatty-acid composition is a crucial aspect of pig meat quality. An increase in polyunsaturated lipid fraction in muscle and fat tissues, although favourable from the standpoint of human nutrition, can be undesirable for technological and sensory qualities of fresh pork and other pig meat products. Lawrence & Fowler (1997) reported that a high content of linoleic acid [C18:2(n-6)] in pig meat is associated with low consumer acceptability. Lipid degradation, mainly through oxidation of free unsaturated fatty acids during the processing of pig meat products, leads to the formation of numerous volatile compounds, some of which contribute to their desirable flavour (Ruiz et al. 1999). However, the presence of an excessive content of PUFA may lead to oxidative rancidity that causes abnormal flavour and softness of the final product (Russo & Nanni Costa 1995; Cava et al. 1999).
The first report of QTL affecting fatty-acid composition identified a significant locus on chromosome 4 (SSC4) for C18:2(n-6) and C18:1(n-9) content in backfat using an Iberian × Landrace pig intercross (IBMAP) (Pérez-Enciso et al. 2000) that was also used in this study. Another genome scan using this pedigree found significant QTL on SSC4, 6, 8, 10 and 12 influencing the content of diverse fatty acids, average chain length (ACL), double bond index (DBI), peroxidability index (PI) and unsaturated index (UI) (Clop et al. 2003). Varona et al. (2004) refined the QTL analysis on SSC6 using the same intercross, and three QTL affecting SFA, MUFA and PUFA were found.
Clop et al. (2003) proposed fatty acid synthase (FASN) and acetyl-CoA carboxylaseα (ACACA) genes as positional and functional candidate genes underlying the QTL on SSC12. The products of both genes play a central role in lipid metabolism as key enzymes in the fatty-acid synthesis (Wakil et al. 1983; Tong 2005). We propose gastric inhibitory polypeptide (GIP) as another positional and functional candidate, based upon its synteny in the comparative human-porcine map and due to its role in the regulation of lipid homeostasis, as demonstrated in several studies for obesity in animals (Meier & Nauck 2004).
The first objective of this work was to detect polymorphisms in the coding regions of porcine FASN, ACACA and GIP, and to map GIP by linkage and physical mapping. Secondly, the previously detected QTL on SSC12 was fine-mapped using a larger sample of animals and increasing the number of genotyped markers. Finally, association analyses between the allelic variants and the fatty-acid profile of backfat tissue were performed to test the contribution of these genes on these traits.
Materials and methods
Animals and phenotypic information
The IBMAP population, previously described by Óvilo et al. (2000), consists of an experimental cross between three Iberian Guadyerbas boars and 30 Landrace sows, with 70 F1 and 403 F2 animals. Fatty-acid composition was measured in the F2 pigs by gas chromatography in backfat samples taken between the third and the fourth ribs. UI was calculated following Pérez-Enciso et al. (2000). ACL, DBI and PI ratios were calculated as described in Pamplona et al. (1998) (Table S1).
Neutral markers genotyping
We added three microsatellites (SW1307, SWR1802 and SWR1021) to the three microsatellites (S0143, SW874 and S0106) and one GH PCR-RFLP marker (HhaI) used in the previous study (Clop et al. 2003). Amplified products were analysed by capillary electrophoresis and fluorescent detection in an ABI 3100 (Applied Biosystems).
Candidate gene analysis
cDNA amplification and sequencing:
Total RNA was isolated from samples of two Iberian (Guadyerbas line) and two Landrace pigs with Tri Reagent (Sigma-Aldrich). First-strand cDNA was synthesized using 5 μg of total RNA, SuperscriptTM II Reverse Transcriptase (Invitrogen) and random hexamers following the supplier's instructions. Standard PCR reactions were performed on a PTC-100 thermocycler (MJ Research) and the RT-PCR products were sequenced in both directions with the Dye-Terminator Cycle Sequencing 3.0 kit in an ABI 377 automatic sequencer (Applied Biosystems). Sequences were edited and aligned with the winstar package to search for SNP.
PCR-RFLP genotyping:
PCR-RFLP protocols were implemented to genotype the whole-resource IBMAP material for the FASN, ACACA and GIP loci. Digestion reactions were performed in 15-μl volumes containing restriction enzyme buffer, 3 U specific enzyme and 5 μl PCR product.
FASN analysis
The FASN gene has not been previously characterised in pig, but has a complex structure in human, with 43 exons spanning more than 19 kb (NM_004104). A search for polymorphisms in the coding region of this gene was performed using backfat cDNA as starting material.
For the amplification of porcine cDNA, several primer pairs were designed from the conserved regions of human, mouse and rat cDNA sequences (NM_004104, NM_007988 and NM_017332 respectively). Porcine FASN cDNA was amplified in 20 overlapping fragments covering exons 2 to 42 (7235 bp) using the primers as shown in Table S2 and sequenced (AY954688). Comparison of Iberian and Landrace sequences allowed the identification of ten SNPs (Table S3). Three missense polymorphisms, located in exons 9 (c.1254A>G), 21 (c.3189T>C) and 39 (c.6545A>C), were genotyped by PCR-RFLP (Table S4).
GIP analysis
The porcine GIP gene had not been previously characterized; however, there is a porcine trace sequence that contains the total cDNA sequence of GIP (AY609506), which was used to design a primer pair (Table S5). A region of 521 bp spanning exon 3 to exon 6 was successfully amplified from duodenum samples and sequenced with approximately 79% identity with human sequence. Comparisons between Iberian and Landrace sequences allowed us to detect three polymorphisms, two of them resulting in amino acid changes (Table S3).
A second primer pair (Table S4) was designed to amplify a ∼350-bp fragment of intron 2 (EF606729) which was used for genotyping the GIPAY609506:c.394A>C SNP. Both GIP missense polymorphisms (c.77A>G and c.394A>C) were genotyped on the pedigree by PCR-RFLP using conditions as given in Table S4. In addition, mapping of GIP was performed using the porcine radiation hybrid panel (ImpRH7000, INRA, Toulouse) and a porcine-specific primer pair amplifying a 99-bp fragment of exon 5 (Table S5). The GIP gene was mapped on SSC12 with retention frequencies of 0.28 in the same linkage group as SS04H11 (LOD score = 19.35).
ACACA analysis
The human ACACA is approximately 330 kb, with 64 exons. The 10-kb mRNA has an open reading frame of 7038 bp that encodes a 265-kDa polypeptide of 2346 amino acids in the longest isoform (56 exons). Because the porcine ACACA gene has not been previously characterized, total RNA was obtained from liver samples so as to carry out sequencing. The complete gene sequence of human (NM_198834), rat (NM_022193) and cattle (NM_174224) ACACA were aligned with a fragment of the porcine sequence (AF252267) and primers were designed from the conserved regions (Table S6). The final sequence from 17 overlapping amplified fragments was submitted to GenBank (EF618729). The analysis of 7020 bp in two Iberian and two Landrace samples allowed the detection of ten polymorphisms. The last 1930 bp were re-sequenced in six additional Landrace pigs and five new polymorphisms were identified (Table S3).
ACACA polymorphisms EF618729:c.5634T>C and EF618729:c.6681G>T in exons 46 and 54 respectively, were genotyped across the pedigree using PCR-RFLP protocols (Table S4).
Statistical analysis
 (1)
(1) (2)
(2) (3)
(3)where yi is the ith individual record, βc is a covariate coefficient with c being carcass weight, a is the QTL additive effect, d is the dominant effect, and βa and βd are the additive and dominant coefficients respectively. Batch was the slaughter batch (nine in total). Sex and batch are considered as fixed effects. We also used model (1) adding backfat thickness, to test whether the QTL were real or attributable to differences in fat deposition. Dominance was not significant for most fatty acids and consequently model (2) only included additive effects. In models (2) and (3), λ was a −1/1 indicator variable depending on whether the individual was homozygous for the alternative alleles (heterozygous animals were given a value of 0, and g represented the allele effect. Finally, u was the infinitesimal genetic effect and e was the residual. The SNP tested in this study were c.5634T>C (in ACACA), c.1254A>G (in FASN), c.77A>G (in GIP) and c.394A>C (in GIP).
Models (1) and (3) were fitted every cM across SSC12. Nominal P-values were obtained assuming a chi-square distribution of the likelihood ratio (LR) test, with degrees of freedom given by the difference between the number of estimated parameters in the reduced and full models. Additional analyses were performed to distinguish between linked and pleiotropic QTL using bivariate models fitting one or two QTL effects. All statistical analyses were carried out with qxpak software under maximum likelihood (Pérez-Enciso & Misztal 2004). Significance thresholds for the QTL were calculated using the procedure described by Nezer et al. (2002). This approach yielded chromosome-wise critical values of LR tests with 2 d.f. of 19.19, 14.86 and 11.29, associated with type I errors of 0.1, 1 and 5% respectively. The corresponding values with 1 d.f. were 15.87, 11.80 and 8.51 respectively. The 95% confidence intervals for QTL locations were calculated according to Mangin et al. (1994).
Results and discussion
Linkage analysis
Genotype information from three additional microsatellites (SW1307, SWR1802 and SWR1021) and from SNPs in three genes (FASN, ACACA and GIP) was added to genotype data from a previous study (Clop et al. 2003) to construct a SSC12 linkage map. All data were in agreement with previous evidence regarding the order and marker distances (http://www.genome.iastate.edu/maps/marcmap.html). The map obtained was FASN– 2.1 –S0143– 29.5 –GH– 2.7 –SW1307– 16.3 –SW874– 7.7 –GIP– 10.8 –SWR1802– 6.4 –ACACA– 20.0 –S0106– 8.5 –SWR1021, with a total length of 111.7 cM in the sex-averaged map. The mean distance between markers was 12.4 cM. FASN, GIP and ACACA were mapped to positions 0, 58.4 and 75.6 cM respectively.
Detection of QTL for fatty-acid contents on chromosome 12
The mean and standard deviation of the phenotypic data are presented in Table S1. LR profiles across SSC12 revealed at least two intervals at positions 11–34 and 68–76 cM respectively, harbouring QTL for diverse fatty acids with maxima above the critical LR values associated with type I errors of 1% (Fig. 1). The main results are detailed in Table 1. The most significant detected QTL for fatty-acid composition affected the percentage of palmitoleic [C16:1 (n-9)] and mapped to 68 cM, between GIP and SWR1802. Nearby, a QTL (75–76 cM) affecting the percentages of both C18:0 and C18:1(n-7) was coincident with the location of the ACACA. The Iberian allele (Q) increased the content of MUFA C16:1(n-9) and C18:1(n-7), and decreased the content of SFA C18:0. Gene action was additive for C18:0 and C18:1(n-7) and dominant for C16:1(n-9), whereas the heterozygous combination (Qq) was closer to the homozygous Landrace genotype (qq).

Likelihood-ratio test profiles across chromosome 12. Horizontal continuous and dashed lines indicate 0.1 and 1% significance threshold respectively.
| Trait | Position in cM (CI) | LR | Nominal P-value | a (SE) | d (SE) |
|---|---|---|---|---|---|
| C14:0 | 11 (3–19) | 14.23** | 2 × 10−4 | 0.04 (0.01) | – |
| C16:0 | 20 (9–32) | 12.64** | 4 × 10−4 | 0.35 (0.10) | – |
| C16:1(n-9) | 68 (63–77) | 17.04** | 2 × 10−4 | 0.09 (0.03) | −0.12 (0.04) |
| C18:0 | 75 (68–84) | 9.35** | 0.002 | −0.26 (0.08) | – |
| C18:1(n-7) | 76 (69–83) | 10.75** | 0.001 | 0.08 (0.02) | – |
| C20:1(n-9) | 34 (22–40) | 12.34** | 4 × 10−4 | −0.05 (0.01) | – |
| C20:2(n-6) | 1 (1–21) | 13.25** | 3 × 10−4 | −0.03 (0.01) | – |
| ACL | 18 (8–27) | 17.27*** | 3 × 10−5 | −0.02 (0.01) | – |
- LR, likelihood ratio; SE, standard error.
- **P < 0.01; ***P < 0.001 (chromosome-wise significance).
We observed significant QTL effects in the genetic interval from 11–34 cM for the percentages of myristic (C14:0), C16:0, gadoleic [C20:1(n-9)] and eicosadienoic [C20:2(n-6)] fatty acids, as well as on ACL. However, the effect on PUFA [C20:2(n-6)] was dependent upon the model used for the statistical analysis. A significant QTL effect for this fatty acid was detected when we used a model adding backfat thickness as a covariate instead of carcass weight. The effects on the other fatty acids [C14:0, C16:0 and C20:1(n-9)] probably correspond to a single QTL, as their confidence intervals partially overlap between 3–40 cM (Table 1 and Fig. 1). Additionally, LR tests values for the comparison between a two-linked QTL model vs. a single pleiotropic QTL model were lower than the nominal value of significance threshold (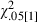 = 3.84) for the different pairs of these fatty acids (results not shown). As a consequence, the null hypothesis of one pleiotropic QTL cannot be rejected. Here the Iberian allele (Q) increased the content of SFA C14:0 and C16:0 and decreased the content of MUFA C20:1(n-9). Gene action was additive for these three fatty acids.
= 3.84) for the different pairs of these fatty acids (results not shown). As a consequence, the null hypothesis of one pleiotropic QTL cannot be rejected. Here the Iberian allele (Q) increased the content of SFA C14:0 and C16:0 and decreased the content of MUFA C20:1(n-9). Gene action was additive for these three fatty acids.
Several studies have been performed in experimental crosses to detect porcine QTL affecting fatty-acid content in fat tissues. QTL on SSC1, 4, 7 and 18 affecting C18:2(n-6), C18:1(n-9) and C14:0 were detected in a resource population, obtained by diverse crosses between Landrace × Yorkshire (Lee et al. 2003; Kim et al. 2006). Recently, Nii et al. (2006) analysed QTL for fatty-acid composition in perirenal fat and backfat using a Japanese × Large White intercross. QTL for diverse fatty acids were mapped in that genome scan on SSC1, 2, 3, 4, 5, 6, 9, 14, 15, 16, 17 and X. However, QTL for fatty acids on SSC12 have only been detected in the Iberian × Landrace cross, which significantly affected the percentage of 18:3 (n-3) fatty acid and showed suggestive effects for the percentages of C18:1(n-7) and C20:1(n-9) fatty acids, as well as ACL and PI (Clop et al. 2003). These effects were attributed to a single QTL mapping on the proximal chromosome interval, between the S0143 and GH markers. The discrepancies between Clop's et al. (2003) study and our study are assignable to the use of six new informative markers and the genotyping of 30 additional F2 pigs.
Candidate gene association analysis
We applied model (2) to those cases where the QTL mapped close to one of the candidate genes; this model is equivalent to standard QTL analysis (model 1) when alternative alleles for each gene were fixed in each parental breed. However, the SNP frequencies in the parental breeds greatly differed from this assumption, and association analysis allows some discrimination.
The FASN gene was evaluated as candidate gene for QTL at 3–40 cM on SSC12 and GIP and ACACA genes for QTL on 68–76 cM. It should be noted that an association analysis of animals in an F2 design is highly confounded with linkage disequilibrium. The model (3) includes the probability of line origin given the neutral markers, and its application (marker-assisted association test) takes into account the between breed linkage disequilibrium. Results of the association analyses using these models are summarized in Tables 2 & 3.
| SNP | Trait | LR | P-value | g (SE)1 |
|---|---|---|---|---|
| FASN: c.1254A>G | C14:0 | 0.52 | 0.471 | −0.02 (0.02) |
| C16:0 | 1.23 | 0.267 | −0.16 (0.15) | |
| C20:1(n-9) | 3.96 | 0.046 | −0.05 (0.02) | |
| ACL | 0.49 | 0.488 | 0.01 (0.01) | |
| GIP: c.77A>G | C16:1(n-9) | 10.54 | 0.001 | 0.12 (0.04) |
| C18:0 | 5.07 | 0.024 | −0.36 (0.11) | |
| C18:1(n-7) | 0.70 | 0.401 | 0.02 (0.03) | |
| GIP: c.394A>C | C16:1(n-9) | 4.87 | 0.027 | 0.06 (0.03) |
| C18:0 | 2.25 | 0.133 | −0.13 (0.08) | |
| C18:1(n-7) | 0.02 | 0.900 | 0.00 (0.02) | |
| ACACA: c.5634T>C | C16:1(n-9) | 14.23 | 8 × 10−4 | 0.07 (0.03) |
| C18:0 | 4.00 | 0.046 | −0.28 (0.08) | |
| C18:1(n-7) | 13.77 | 2 × 10−4 | 0.09 (0.02) | |
| ACACA: c.6681G>T | C16:1(n-9) | 3.11 | 0.078 | −0.05 (0.03) |
| C18:0 | 9.00 | 0.003 | 0.24 (0.08) | |
| C18:1(n-7) | 0.97 | 0.325 | −0.02 (0.02) |
- LR, likelihood ratio test; SE, standard error.
- 1Additive effect with SE.
| SNP effect | QTL | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| SNP | Trait | LR1 | P-value | g (SE)2 | Position (cM) | LR1 | Nominal P-value | a (SE)3 | d (SE)3 |
| FASN: c.1254A>G | C20:1(n-9) | 1.95 | 0.162 | −0.03 (0.02) | 34 | 10.30* | 0.001 | −0.04 (0.01) | – |
| GIP: c.77A>G | C16:1(n-9) | 11.86 | 6 × 10−4 | 0.12 (0.04) | 68 | 16.61** | 2 × 10−6 | 0.08 (0.03) | −0.12 (0.04) |
| C18:0 | 15.52 | 8 × 10−5 | −0.35 (0.10) | 64 | 8.51* | 3 × 10−5 | −0.32 (0.10) | – | |
| GIP: c.394A>C | C16:1(n-9) | 14.27 | 2 × 10−4 | 0.11 (0.03) | 68 | 26.49*** | 2 × 10−8 | 0.13 (0.03) | −0.12 (0.04) |
| ACACA: c.5634T>C | C16:1(n-9) | 12.59 | 4 × 10−5 | 0.12 (0.04) | 70 | 6.05 | 0.048 | 0.05 (0.03) | −0.09 (0.04) |
| C18:0 | 4.09 | 0.043 | −0.32 (0.10) | 58 | 1.09 | 0.297 | 0.10 (0.10) | – | |
| C18:1(n-7) | 12.54 | 4 × 10−4 | 0.08 (0.02) | 0 | 2.75 | 0.097 | 0.03 (0.02) | – | |
| ACACA: c.6681G>T | C18:0 | 8.48 | 0.004 | 0.24 (0.08) | 48 | 0.32 | 0.573 | −0.05 (0.08) | |
- 1Likelihood ratio test at chromosome-wise significance: *P < 0.05; **P < 0.01 and ***P < 0.001.
- 2Additive effect of the corresponding SNP with SE, standard error.
- 3Additive effect and dominance effect of QTL with SE.
We tested FASN as a candidate gene underlying the QTL affecting ACL, C14:0, C16:0 and C20:1(n-9) fatty acids. FASN encodes for a central multifunctional enzyme that is the responsible of de novo fatty-acid biosynthesis (Roy et al. 2001) and had been located by physical and linkage mapping on SSC12 (Muñoz et al. 2003). Morris et al. (2007) found five SNPs in bovine FASN significantly associated with fatty-acid percentage in adipose fat or milk fat. These results suggested strong linkage disequilibrium between these SNPs and the causal mutation. In our analysis, sequence comparisons of a fragment of 7235 bp of the porcine FASN cDNA from Iberian and Landrace pig breeds allowed detection of three missense SNPs that were cosegregating in two haplotypes of FASN (c.[1254A; 3189T; .6545A] and c.[1254G; 3189C; 6545C]). The first was fixed in the Iberian parental population and had a frequency of 0.85 in Landrace, while the second was only present in Landrace at 0.15. Frequencies of these haplotypes in F2 pigs were 0.91 and 0.09 respectively. The FASN c.1254A>G SNP was only associated with the percentage of C20:1[n-9] fatty acid, with a negative effect of the A allele, which had frequencies of 1.0 and 0.85 in the parental lines. However, the result of the correspondent marker-assisted association test (Table 3) indicated that the QTL effect was more significant than the SNP (nominal P-values = 0.001 and 0.162 respectively). The three genotyped SNPs produced amino acid changes (Arg418Gln, Thr1063Ile and His2182Asn), but differences in the protein structure were not found due to these changes by the structure prediction tool ‘Simple Modular Architecture Research Tool’ (SMART) (http://smart.embl-heidelberg.de) (Letunic et al. 2006). Additionally, the amino acid changes were estimated as tolerated for the protein function by Sorting Intolerant From Tolerant tool (sift); http://blocks.fhcrc.org/sift/SIFT.html) (Ng & Henikoff 2001). Therefore, there was no indication that the detected intragenic haplotypes in the coding region of the FASN gene explained the effects of the first QTL.
Acetyl-CoA oxidase 1 (ACOX1) and ATP citrate lyase (ACLY) constitute potential candidate genes for this QTL. ACOX1 encodes for a protein that is the first enzyme of the fatty-acid beta-oxidation pathway, which catalyses the desaturation of acyl-CoA to 2-trans-enoyl-CoA. ACLY encodes for the primary enzyme responsible for the synthesis of cytosolic acetyl-CoA in many tissues, which serves several important biosynthetic pathways, including lipogenesis and cholesterogenesis. These genes have not been mapped in pigs, but human–pig comparative map shows that human localization for the ACOX1 and ACLY genes (17q24-q25 and 17q1-q21 respectively) could match to the first QTL interval detected in our study.
The GIP gene was evaluated as a candidate gene responsible for the QTL effects on C16:1(n-9), C18:0 and C18:1(n-7) fatty acids. GIP plays a physiological role in fat accumulation in adipose tissues (Yamada et al. 2006). A region of 521 bp of the cDNA was successfully characterized and three SNPs were detected. Results of GIP mapping showed that both linkage and physical maps agreed in the position of the gene as well as the prediction from the comparative human–porcine map. Both missense SNPs were genotyped across the pedigree. The GIP:c.77A allele had frequencies of 0.33 and 0.13 frequency values in the parental Iberian and Landrace animals respectively, while the GIP c.394A allele had 0.17 and 0.67 frequencies respectively. Both polymorphisms were arranged in four haplotypes in the F2 pigs: c.[77A; 394A], c.[77A; 394C], c.[77G; 394A] and c.[77G; 394C]. Haplotype frequencies were 0.14, 0.06, 0.25 and 0.55 for the F2 pigs for which haplotypes could be assigned. A significant association was observed between the GIP polymorphism c.77A>G and the percentages of C18:0 and C16:1(n-9). The effect of the c.77A allele was positive on C16:1(n-9) and negative on C18:0. The other tested GIP polymorphism c. 394A>C was also significantly associated with C16:1(n-9) but to a lesser magnitude (Table 2). The results of marker-assisted association tests showed highly significant effects for both SNPs and QTL (Table 3). Both tested SNPs produce amino acid changes (Asp26Gly and Met132Leu respectively); however, no differences in the protein structure due to the alternative alleles were predicted using SMART, and in the same way, the amino acid changes were estimated as tolerated using sift. Both association and function prediction analyses suggested that the tested polymorphisms were not causal but may be in linkage disequilibrium with the mutation.
Similarly, ACACA constitutes a good positional and functional gene responsible for the QTL effect on C16:1(n-9) and C18:0 and C18:1(n-7) fatty acids. Acetyl-CoA carboxylase catalyses the carboxylation of acetyl CoA to malonyl CoA, the rate-limiting enzyme for long-chain fatty-acid synthesis and it was previously mapped on SSC12 (Calvo et al. 2000). Almost the complete porcine cDNA (7020 bp) was successfully characterized and the sequence comparisons revealed 15 synonymous SNPs, two of them were genotyped across the pedigree. The ACACA:c.5634C allele was fixed in the Iberian parents and presented frequencies 0.43 and 0.69 in the Landrace and F2 pigs respectively. The alleles of the silent ACACA c.6681G>T polymorphism presented intermediate frequencies. Four haplotypes were found in the F2 generation from the combination of these SNP: c.[5634C; 6681T], c.[5634C; 6681G], c.[5634T; 6681T] and c.[5634T; 6681G]. Haplotype frequencies were 0.25, 0.45, 0.23 and 0.01 respectively. Significant associations were found between the ACACA c.5634T>C SNP and the three analysed fatty acids with models 2 and 3 (Tables 2 & 3), with a negative effect of the c.5634C allele on the percentage of C18:0, and positive on C16:1(n-9) and C18:1(n-7). The ACACA c.6681G>T polymorphism was significantly associated with C18:0, with a positive effect of the c.6681G allele. Under model (3), the QTL for the fatty-acid affected by the two ACACA mutations was not significant, and two out of them mapped on a different location (Table 3). As none of the 15 detected SNPs produced amino acid changes, no candidate mutation could be tested. However, the positive results of the association analyses indicate that the synonymous mutations in ACACA, c.5634T>C and c.6681G>T, would be in linkage disequilibrium with the causal mutation.
In the present study, the affected SFA (C18:0) and MUFA [C16:1(n-9) and C18:1(n-7)] were not related with an increase in cardiovascular disease risk. Moreover, the association results were obtained using an experimental cross, the magnitude of the estimated effects were low and the affected fatty acids did not include the concentration of oleic acid, which is the most abundant and nutritionally relevant fatty acid (López-Bote 1998; Lo Fiego et al. 2005; Lichtenstein 2006). For all these reasons, we do not encourage using these GIP and ACACA polymorphisms as linked markers in marker-assisted selection to enhance genetic changes on fatty-acid composition.
Acknowledgements
We are grateful to Miguel Pérez-Enciso and Ignacy Misztal for the qxpak software and Nines López for technical assistance. We are indebted to Nova Genética (Lleida, Spain) and SIA ‘Dehesón del Encinar’ (Oropesa, Spain) and their staff for providing the experimental pigs. We thank Dr Milan (INRA) for providing the ImpRH panel. A. Fernández is funded by a fellowship from Instituto Nacional de Tecnología Agraria y Alimentaria (INIA), and J. Estellé is supported by a fellowship from the Ministerio de Educación y Ciencia (MEC). This work was funded by INIA grant CPE-010-C3.

