

Dermokine contributes to epithelial–mesenchymal transition through increased activation of signal transducer and activator of transcription 3 in pancreatic cancer
Funding Information:
Innovative Research Groups of the General Surgery of Wenzhou, Zhejiang, China (Grant/Award Number: No. C20150003), Provinces and Ministries Co-Contribution of Zhejiang, China (Grant/Award Number: No. wkj-zj-1706)
Abstract
Dermokine (DMKN) was first identified in relation to skin lesion healing and skin carcinoma. Recently, its expression was associated with pancreatic cancer tumorigenesis, although its involvement remains poorly understood. Herein, we showed that DMKN loss of function in Patu-8988 and PANC-1 pancreatic cancer cell lines resulted in reduced phosphorylation of signal transducer and activator of transcription 3, and increased activation of ERK1/2 and AKT serine/threonine kinase. This decreased the proliferation ability of pancreatic ductal adenocarcinoma (PDAC) cells. In addition, DMKN knockdown decreased the invasion and migration of PDAC cells, partially reversed the epithelial–mesenchymal transition, retarded tumor growth in a xenograft animal model by decreasing the density of microvessels, and attenuated the distant metastasis of human PDAC in a mouse model. Taken together, these data suggested that DMKN could be a potential prognostic biomarker and therapeutic target in pancreatic cancer.
Pancreatic ductal adenocarcinoma (PDAC) is the fourth leading cause of cancer-related deaths in the USA.1 It is usually diagnosed at an advanced stage, because early-stage cancer is mostly asymptomatic and appropriate serological biomarkers have not been established. Accordingly, the prognosis for advanced pancreatic cancer remains very poor. Early metastasis and local invasion are the main reasons for the high mortality; approximately 85% of patients are diagnosed at an unresectable stage.2 Therefore, it is of high importance to identify novel biomarkers that would improve early diagnosis and therapeutic outcomes in pancreatic cancer.
DMKN, a gene located at human chromosome 19q13.12, encodes 10 putative dermokine (DMKN) transcript isoforms in normal epidermis.3 DMKN is upregulated in inflammatory skin disorders, but downregulated in skin cancer. Higashi et al.4 reported that dermokine-β could impair phosphorylation of ERK in keratinocytes or skin tumor cells. Recent studies have detected DMKN in malignant tumors of the digestive tract, including invasive pancreatic carcinoma and colorectal cancer.5 Basciano et al.6 showed that DMKN was upregulated approximately 10-fold in mesenchymal stem cells after long-term culture under hypoxic conditions. Interestingly, our previous study suggested that hypoxia was one of the factors contributing to pancreatic cancer progression.7 Therefore, we hypothesized a link between DMKN and pancreatic cancer. At present, neither upstream nor downstream targets of DMKN related to malignancy of pancreatic cancer have been identified.
Approximately 90% of pancreatic cancers correspond to PDACs, with approximately 95% of patients presenting mutations in K-Ras GTPase (KRAS). Once mutated, KRAS remains in a continuously active state, interacting with diverse downstream effectors and signaling pathways associated with growth, invasion, and metastasis.8, 9 Targeting of the downstream effectors of KRAS, particularly the MAPK and PI3K-AKT signaling pathways, represents a promising strategy to prevent PDAC progression.10 Signal transducer and activator of transcription 3 (STAT3) is a transcription factor that is a member of the STAT protein family encoded by the STAT3 gene. Activation of STAT3 may occur through phosphorylation by MAPK11 and PI3K12 signaling pathways. Several genes and signaling molecules associated with these pathways are closely related to PDAC growth, invasion, and metastasis, including cell-cycle and proliferation regulators, tumor angiogenesis agonists, and epithelial–mesenchymal transition (EMT) mediators.9
Here, we detected DMKN protein and mRNA in pancreatic cancer tissue samples and cell lines, respectively. We also assessed the link between DMKN expression and patient tumor stage, as well as the effect of DMKN silencing on PDAC proliferation and metastasis in vitro. Furthermore, in vivo assays suggested that PDAC cells with downregulated DMKN attenuated xenograft growth in nude mice. Finally, our work provides evidence that DMKN affects the activation of STAT3 and downstream molecular proteins of the MAPK and PI3K signaling pathways, which may provide insights into both the mechanisms underlying carcinogenesis and potential biomarkers for PDAC.
Materials and Methods
Cell lines and cell culture
The human pancreatic cancer cell lines PANC-1, BxPC-3, and Patu-8988 as well as HDPE6C7 immortalized pancreatic duct epithelial cells were obtained from the Chinese Academy of Sciences Cell Bank (Shanghai, China) and cultured in either DMEM or RPMI-1640 medium supplemented with 10% FBS (all Gibco, Thermo Fisher Scientific, Waltham, MA, USA). Cells were maintained at 37°C in 5% CO2.
Patients and specimens
The clinical research protocol was approved by the Ethical Committee of the First Affiliated Hospital of Wenzhou Medical University (Wenzhou, China). In total, 44 patients diagnosed with pancreatic cancer who had undergone pancreatic resection to cure early pancreatic cancer or reduce tumor occupancy of advanced pancreatic cancer were enrolled. Tissue specimens were collected and, following surgery, pancreatic adenocarcinoma was histopathologically diagnosed and confirmed by the pathology department, according to the criteria of the WHO. Patient details are listed in Table 1.
| Clinicopathologic variables | n | DMKN† | P-value |
|---|---|---|---|
| Gender | |||
| Male | 32 | 13.75 (9.56–19.27) | 0.687 |
| Female | 12 | 11.14 (9.65–18.26) | |
| Age, years | |||
| <60 | 8 | 11.22 (7.82–19.68) | 0.360 |
| ≥60 | 36 | 13.75 (10.01–19.26) | |
| Differentiation | |||
| Poorly or none | 28 | 13.68 (8.82–19.02) | 0.696 |
| Well or moderately | 16 | 13.03 (9.65–19.10) | |
| Primary tumor‡ | |||
| T1/2 | 13 | 10.95 (7.18–15.41) | 0.030 * |
| T3/4 | 31 | 15.39 (11.11–19.77) | |
| Regional lymph nodes‡ | |||
| N0 | 30 | 14.26 (10.74–18.54) | 0.597 |
| N1 | 14 | 12.58 (8.83–20.29) | |
| Distant metastasis‡ | |||
| M0 | 14 | 11.06 (7.71–14.60) | 0.028 * |
| M1 | 30 | 16.25 (10.88–19.86) | |
- *P-value <0.05 (shown in bold). †DMKN expression is shown as protein integrated optical density ×10−4; median of relative expression in the 25th–75th percentile is listed in parentheses. ‡According to UICC/AJCC Guideline Version 7 Pancreatic Cancer staging.
RNA isolation and quantitative real-time PCR
Total RNA was extracted from tumor cells using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. cDNA synthesis was carried out using the Revert Aid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific). Quantitative real-time PCR (qRT-PCR) was carried out on a 7500 Real-Time PCR Detection System (Applied Biosystems, Foster City, CA, USA) using the SYBR-Green Master Mix kit (Roche, Indianapolis, IN, USA) under standard conditions following the manufacturer's instructions. GAPDH was used as an internal control and results were analyzed using the 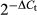 method. The primers used were listed in Table 2.
method. The primers used were listed in Table 2.
| Gene | Forward primer (5′–3′) | Reverse primer (5′–3′) |
|---|---|---|
| GAPDH | tgacttcaacagcgacaccca | caccctgttgctgtagccaaa |
| DMKN-α | ccactgcctctgctctgc | gttgtaattgtagttctgatcggct |
| DMKN- β/γ | ggcacgagattggcagacag | ccagaagtttcccaagcacc |
| DMKN-γ | aactgggatgccataaacaag | aagggctgagattagtgatga |
| CDH-1 | tcacgctgtgtcatccaacgg | taggtgttcacatcatcgtccgc |
| CDH-2 | catcatcatcctgcttatccttgt | ggtcttcttctcctccaccttctt |
| SNAI1 | cttctcctctacttcagtctcttcc | tgaggtattccttgttgcagtattt |
| Vimentin | ggatgttgacaatgcgtctc | ctcctggatttcctcttcgt |
| VEGF | ggaggcagagaaaagagaaagtgt | aagagagcaagagagagcaaaagat |
| MMP-2 | gggggaagatgctgctgtt | agcggtcctggcagaaatag |
| MMP-9 | cagtccacccttgtgctcttcc | catctctgccacccgagtgtaac |
Transduction with DMKN shRNA
Human DMKN shRNA lentivirus was purchased from Gene Chem (Shanghai, China). One day before transduction, cells were plated at a density of 5 × 105 per well in six-well plates containing 2 mL medium. After culturing for 24 h, the cells reached 40% confluence and the medium was replaced with 1 mL medium containing lentiviral particles plus 10 μg/mL polybrene, according to the manufacturer's instructions. After incubation for another 12 h, lentivirus-containing medium was replaced with fresh medium. Transfection efficiency was evaluated using qRT-PCR and Western blot analysis.
Transfection with DMKN plasmid
The human DMKN plasmid was designed according to the sequence of NM_033317.4, the longest transcript that encodes the longest isoform of DMKN, also referred to as isoform beta. One day before transfection, cells were plated at a density of 1 × 106 per well in six-well plates containing 2 mL medium. After culturing for 24 h, the cells reached 50% confluence and the medium was replaced with 2 mL complete DMEM containing 3 μg/mL empty plasmid or DMKN plasmid. Subsequent experiments were carried out after incubation for another 24 h.
Tumor xenograft assay
PANC-1 cells (2 × 106) transduced with DMKNβ/γ shRNA (knockdown [KD] group) or GFP (normal control [NC] group) lentivirus were resuspended in 100 μL culture medium without FBS and injected s.c. into 4-week-old female (BALB/c) nude mice (n = 5/group). The mice were purchased from SLAC Co. Ltd. (Shanghai, China) and raised under specific pathogen-free conditions. All animal procedures were approved by the Ethical Committee of Wenzhou Medical University and Laboratory Animal Management Committee of Zhejiang Province (Approval ID: wydw2015- 0146). Tumor volume was calculated as (length × width2)/2 and measured twice a week. The mice were killed after 4 weeks.
Tumor metastasis assay in vivo
PANC-1 cells (2 × 106) transduced with DMKN shRNA (KD group) or GFP (NC group) lentivirus were resuspended in 100 μL culture medium without FBS and i.v. injected into the tail vein of 4-week-old female (BALB/c) nude mice (n = 7/group) raised under specific pathogen-free conditions. Tumor metastasis was screened using an IVIS Lumina II system (Caliper Life Sciences, Boston, MA, USA) and analyzed using Living Image software (PerkinElmer Health Sciences, Waltham, MA, USA).
Western blot analysis
Western blots were carried out according to standard procedures. Total protein was extracted with RIPA buffer (Beyotime Biotechnology, Shanghai, China) in the presence of PMSF (Beyotime Biotechnology) and PhosSTOP (Roche). The following commercially available antibodies were used: anti-GAPDH (Bioworld Technology, St. Louis Park, MN, USA), anti-DMKN (Abcam, Cambridge, MA, USA), anti-vascular endothelial growth factor (VEGF; Abcam), anti-ERK1/2, anti-phospho-ERK1/2, anti-AKT, anti-phospho-AKT, anti-STAT3, anti-phospho-STAT3, anti-E-cadherin, anti-N-cadherin, anti-Snail, anti-vimentin, anti- MMP2, and anti-MMP9 (all Cell Signaling Technology, Danvers, MA, USA).
Proliferation, cell migration, and invasion assays
Patu-8988 and PANC-1 cells transfected with either DMKNβ/γ shRNA or GFP lentivirus were plated in 16-well E-plates at 2 × 104 per well. Growth was monitored continuously for 72 h by measuring impedance using the RTCA SP xCELLigence system (Roche). For the cell migration assay, 8 × 104 cells per well were plated in 16-well cell invasion/migration (CIM) plates and the cell index was monitored continuously for 24 h using the RTCA SP xCELLigence system. For the invasion assay, cells were seeded onto filters in a 24-well Transwell chamber coated with Matrigel (diluted 1:6; BD Biosciences, Franklin Lakes, NJ, USA). Cell migration through the Matrigel substrate was assessed after 24 h by staining with crystal violet and counting under bright-field microscopy.
Cell cycle analysis
The cell cycle was measured by flow cytometry using the Cell Cycle Staining Kit (Multiscience, Hangzhou, China), according to the manufacturer's instructions. Data were analyzed with ModFit LT2.0 software (Verity Software House, Topsham, ME, USA).
Microarray analysis
Total RNA was isolated as described above and the quality was measured with a NanoDrop 2000 system (Thermo Fisher Scientific) and Agilent 2100 Bioanalyzer (Agilent, Palo Alto, CA, USA). Target RNA preparation was undertaken with the GeneChip 3′ IVT Express Kit (Affymetrix, Santa Clara, CA, USA). Samples were hybridized to the GeneChip PrimeView Human Gene Expression Array following the manufacturer's instructions. Microarrays were then washed and stained with GeneChip Hybridization Wash and Stain Kit and scanned with the GeneChip Scanner 3000. Data were analyzed with Ingenuity Pathway Analysis (Qiagen, Redwood City, CA, USA). All analyses were performed with three technical and biological replicates.
Immunohistochemical staining
Paraformaldehyde-fixed paraffin-embedded sections of human and nude mice tumor tissue were subjected to immunohistochemical staining (IHC) following standard protocols. The following antibodies were used: anti-DMKN (Santa Cruz Biotechnology, Dallas, TX, USA) and anti-CD31 (Abcam). The integrated optical density of DMKN by IHC was analyzed with Image Pro Plus 6.0 (Media Cybernetics, Rockville, MD, USA). Representative images were captured with a Leica DM4000B microscope (Leica, Jena, Germany).
Statistical analysis
Data were analyzed with spss 22.0 (IBM, Armonk, NY, USA) and Prism 5.0 software (GraphPad Software, San Diego, CA, USA). Data were reported as the mean ± SEM. Analysis of variance and Student's t-test for repeated data were carried out to assess differences between groups. The Mann–Whitney U-test was applied to analyze the correlation between the integrated optical density of DMKN and clinicopathologic characteristics. P < 0.05 was considered statistically significant (*P < 0.05, **P < 0.01, ***P < 0.001).
Results
DMKN was deregulated in pancreatic cancer cell lines and correlated with tumor grade in pancreatic cancer patients
Previous reports indicated that DMKN was absent from most human organs,13 including the normal epithelium of hyperplastic mucosa of the pancreatic duct. Watanabe et al.14 reported that normal pancreatic duct and mucus cell hyperplasia in the main pancreatic duct tissue did not express DMKN. To detect the expression of DMKN in pancreatic cancer, we designed three primers for qPCR based on the mRNA sequence from NCBI (Gene ID: 93099), covering DMKNα, β, and γ. To understand the function of DMKN and its relationship with malignancy in pancreatic cancer, we analyzed DMKN mRNA and protein expression in duct epithelial cells of the pancreas (HPDE 6C7) and three other types of pancreatic cancer cell lines. Both DMKN mRNA and total DMKN protein were upregulated in the pancreatic cancer cell lines (Fig. 1A,B). Additionally, we assessed DMKN protein levels in PDAC samples from 44 patients at different tumor stages (Table 1). Immunohistochemical staining showed that expression of DMKN was associated with tumor invasion (P = 0.035) and cancer metastasis (P = 0.033). Expression of DMKN was significantly higher in T3/4 compared to that in T1/2 tumors, and in M1 compared to that in M0 distant metastasis tumors (Fig. 1C–E). However, no obvious relationship was found between DMKN expression and sex, age, tumor differentiation, or lymphatic metastasis (Table 1).

DMKN silencing slightly inhibited proliferation and induced cell cycle arrest of PDAC cells by altering cell cycle-related genes
Considering the lack of a common sequence region between DMKNα and DMKNβ/γ, we designed shRNA only targeting DMKNβ/γ. To explore the role of DMKNβ/γ in the malignant behavior of pancreatic cancer, we used shRNA to interfere with its expression in pancreatic cancer cells. To avoid off-target effects of the shRNA, we used three types of shRNA-expressing lentivirus targeting DMKNβ/γ mRNA in Patu-8988 cells in our preliminary experiment, and the interference effects of shRNA-DMKNβ/γ were evaluated by qRT-PCR (Fig. 2A-a). The sequences of shRNA-DMKNβ/γ were listed in Table 3. We chose the most effective shRNA-DMKNβ/γ (#3) to apply to Patu-8988 cells and PANC-1 cells in the following experiments. Expression of DMKN protein was nearly completely lost in DMKN-shRNA-transduced cells compared to that in wild-type and control lentivirus-transduced cells, as determined by Western blotting (Fig. 2A-b), and this correlated with lower proliferation rates (Fig. 2B). The DMKN-shRNA-transfected cells showed cell cycle deregulation and frequent arrest in G0/G1 (Fig. 2C,D).

| No. | 5′ | Stem | Loop | Stem | 3′ |
|---|---|---|---|---|---|
| DMKN-RNAi(#1)-a | Ccgg | caTCAACTGGGATGCCATAAA | CTCGAG | TTTATGGCATCCCAGTTGATG | TTTTTg |
| DMKN-RNAi(#1)-b | aattcaaaaa | caTCAACTGGGATGCCATAAA | CTCGAG | TTTATGGCATCCCAGTTGATG | |
| DMKN-RNAi(#2)-a | Ccgg | ggATGTTTAACTTTGACACTT | CTCGAG | AAGTGTCAAAGTTAAACATCC | TTTTTg |
| DMKN-RNAi(#2)-b | aattcaaaaa | ggATGTTTAACTTTGACACTT | CTCGAG | AAGTGTCAAAGTTAAACATCC | |
| DMKN-RNAi(#3)-a | Ccgg | agACGTCTCCTGGGATGTTTA | CTCGAG | TAAACATCCCAGGAGACGTCT | TTTTTg |
| DMKN-RNAi(#3)-b | aattcaaaaa | agACGTCTCCTGGGATGTTTA | CTCGAG | TAAACATCCCAGGAGACGTCT |
To determine whether DMKN was involved in cell cycle progression, we used GeneChip scanning to assess expression patterns in lentivirus control and DMKN-shRNA-transfected PANC-1 cells. As expected, this yielded several cell cycle-associated genes (Fig. 3).

Reduced DMKN expression inhibited migration and invasion of pancreatic cancer cell lines
Normalized GeneChip results showed that 143 genes were upregulated and 380 genes were downregulated between control lentivirus-transduced cells and DMKN-shRNA-transduced cells (Fig. 3A). Among them, results indicated that changes in EMT-associated genes (N-cadherin) following DMKN knockdown (Fig. 3B). We assessed some other EMT-associated genes that were not detected by GeneChip scanning using RT-PCR (Fig. 3C), and Western blot analysis confirmed the differential expression of different EMT proteins (Fig. 4A). Expression of E-cadherin increased, and that of N-cadherin, Snai1, and vimentin decreased. No significant change in the expression of desmoplakin, claudins, or GLI family zinc finger 1 (GLI1) in pancreatic cancer was detected between the NC and KD cells in the normalized GeneChip results (Table S1). Cells with overexpression and reduced expression of DMKN showed no obvious morphological changes (Fig. S1). Functionally, cells with reduced DMKN expression also showed weaker cell migration (Fig. 4B) and lower invasion potential (Fig. 4C,D). Therefore, we suggested that DMKN might enhance the potential for migration and invasion in pancreatic cancer cells by regulating partial EMT-associated proteins.

Knockdown of DMKN suppressed human tumor growth and metastasis in vivo
To evaluate the role of DMKN in cancer promotion in vivo, we s.c. transplanted stable DMKN-shRNA-transduced PANC-1 cells (KD) or control cells (NC) in nude mice, establishing a xenograft animal model. Xenograft tumors were observed 14 days after PDAC cells were seeded in mice; their growth was monitored every other day for the next 14 days (Fig. 5A). As shown in Figure 5(B), tumor volumes in the NC control group were larger than those in the KD group. This effect might be explained by the significantly decreased expression of angiogenesis-related proteins and mRNA, such as VEGF or MMPs, in DMKN-shRNA-transduced PANC-1 cells (Fig. 5C,D). Moreover, based on IHC for CD31, a marker of vascular endothelial cells, resected tumor tissue showed lower microvessel density in the KD group than in the control group (Fig. 5E).

Additionally, the other two groups of mice received a tail vein injection of either stable DMKN-shRNA-transduced PANC-1 cells (KD) or control cells (NC), establishing an animal model of metastasis. Tumor metastasis status was measured using a Caliper IVIS Lumina II system 2 weeks after the tail vein injection. The fluorescence intensity results indicated that the cells with reduced DMKN expression showed weakened metastasis potential compared with those in the control group.
Analysis of the oncogenic pathway regulated by DMKN
Next, we evaluated the effect of transfection with DMKN-shRNA lentivirus on three main oncogenic signaling pathways in PDAC cells. Interestingly, although the levels of the EMT and cell cycle-associated proteins previously identified (Fig. 3) and reported as downstream of MAPK/ERK1/2 and PI3K/AKT decreased, phosphorylation of the same signaling proteins was higher in the KD than in the NC group (Fig. 6A). To test whether DMKN acted as a regulator of STAT3 and downstream molecular target of these two abovementioned oncogenic pathways, we treated PDAC cells with U0126 (an MAPK/ERK kinase inhibitor) and MK-2206 2HCL (an AKT inhibitor). The treatment decreased DMKN protein expression and partially reversed EMT (Fig. 6B), suggesting that DMKN could indeed be the target of these two signaling pathways. Finally, STAT3 activation decreased when DMKN was downregulated (Fig. 6A), whereas treatment with STA-21 (a STAT3 inhibitor) led to an increase in DMKN, phospho-ERK1/2, and phospho-AKT levels (Fig. 6B), suggesting that STAT3 signaling could be the target of DMKN, which was involved in the partial EMT phenomenon. To confirm our results, we treated PANC-1 cells with DMKN plasmids (OE) and empty plasmids (Empty). Overexpression of DMKN resulted in decreased activity of ERK1/2 and AKT, but increased the phosphorylation level of STAT3, and changed the expression of EMT-associated proteins (Fig. 6C). Therefore, we speculated that DMKN could modulate the ERK1/2/STAT3/EMT and AKT/STAT3/EMT signaling pathways.

Discussion
Dermokine was initially found in skin disorders and was considered an epithelial development marker.15 However, recent studies have detected DMKN in various cancer tissues. We previously showed that DMKN was upregulated in trichostatin A-resistant PANC-1 cells. This was accompanied by upregulation of MMP and vimentin genes, which might promote metastasis, suggesting that DMKN could be a malignancy-associated gene.16
Mounting evidence indicated that oncogenic MAPK and PI3K signaling could modulate the malignant biological behavior of PDAC cells, even though the underlying mechanism remains unclear.17-19 Although many in vivo and/or in vitro studies have shown that activated MAPK/ERK1/2 and PI3K/AKT signaling contributed to pancreatic cancer initiation and progression by exacerbating drug resistance,20 distant tumor metastasis, or EMT, the possible regulatory effects of these oncogenic pathways require further research.21, 22 To the best of our knowledge, this was the first report of a molecular link between DMKN and oncogenic pathways in pancreatic cancer cells. Furthermore, we used a loss-of-function assay to confirm that DMKN feedback activated the MAPK and PI3K signaling pathways at the mRNA and protein levels. The upregulation of other tumor-associated downstream targets of these two pathways following DMKN-shRNA transfection suggested that MAPK, PI3K, and STAT3 signaling converge on DMKN. Dermokine levels gradually increased during PDAC progression, whereas negative DMKN expression predicted an optimistic pancreatic cancer clinical stage.
Epithelial–mesenchymal transition is activated in most cancer cells, where it is linked to dissociation from the primary tumor and intravasation into blood vessels, enabling the dissemination of carcinoma cells.23 Reverse EMT therapeutics are being considered as a method to decrease migration and invasiveness of malignant tumors.24, 25 Furthermore, STAT3 has been identified as a central regulator of tumor metastasis.26 Signal transducer and activator of transcription 3 is activated by numerous cytokine receptor-associated tyrosine kinases, growth factor receptors with intrinsic tyrosine kinase activity, and oncogenic proteins;27-30 making STAT3 signaling central to the regulation of cancer invasion and metastasis. Moreover, our previous study showed that several genes downstream of the activated STAT3 pathway were closely related to EMT.31 In the present study, DMKN-shRNA-transfected PDAC cells showed lower expression of N-cadherin, vimentin, and Snail, but higher expression of E-cadherin. At the same time, STAT3 activity also modulated the level of DMKN. Additionally, expression of EMT-associated proteins was partially altered and that of DMKN decreased in PDAC cells treated with U0126 and MK-2206 2HCL. These results suggest that DMKN may control EMT, and play a vital role in regulating MAPK/ERK1/2, PI3K/AKT, and STAT3 signaling.
High density of blood vessels is an indicator of poor prognosis in a variety of tumors.32 Cancer cells secrete many angiogenesis-promoting proteins such as VEGF into the tumor microenvironment.33 Increased VEGF or microvessel density is associated with poor patient outcome.34 Furthermore, MMPs play critical roles in cancer invasion and metastasis by digesting the basement membrane and ECM.35 Xiang et al.36 reported that expression of MMP2/9 was positively correlated with microvessel density in pancreatic cancers. Pancreatic ductal adenocarcinoma requires sufficient levels of nutrients and oxygen to avoid becoming growth-limited and relies on angiogenesis to drain away toxic byproducts released by cancer cells. Therefore, targeting angiogenesis by downregulating VEGF and MMPs in PDAC could retard tumor progression. In our experiments, VEGF levels were significantly reduced in DMKN-shRNA-transfected pancreatic cancer cell lines, at both the mRNA and protein levels. Elevated MMP2 and MMP9 levels were accompanied by increased DMKN levels and tumor xenograft volume. We suggested that DMKN could promote malignancy and angiogenesis of pancreatic cancer cells by increasing the expression of VEGF and MMPs.
Cell proliferation is tightly regulated by cell cycle genes.37-44 In our experiment, however, the negative regulation of DMKN on PDAC cells was not remarkable and the cells showed arrest in G0/G1, warranting further investigation of the mechanism underlying this deregulation.
Although DMKN silencing in pancreatic cancer cells was not completely effective, DMKN was also a key factor associated with PDAC cell proliferation, migration, and invasion in vitro. The effect of DMKN on pancreatic cancer growth in a tumor xenograft mouse model was most significant when tumor volume was measured. Our study indicated that DMKN could serve as a suitable biomarker and novel therapeutic target for pancreatic cancer.
Acknowledgments
The work was supported by the funds of Provinces and Ministries co-contribution of Zhejiang, China [No. wkj-zj-1706] and Innovative Research Groups of the General Surgery of Wenzhou, Zhejiang, China [No. C20150003].
Disclosure Statement
The authors have no conflict of interest.

