

A genome-wide association study for fat-related traits computed by image analysis in Japanese Black cattle
Abstract
The objective of this study was to identify genomic regions associated with fat-related traits using a Japanese Black cattle population in Hyogo. From 1836 animals, those with high or low values were selected on the basis of corrected phenotype and then pooled into high and low groups (n = 100 each), respectively. DNA pool-based genome-wide association study (GWAS) was performed using Illumina BovineSNP50 BeadChip v2 with three replicate assays for each pooled sample. GWAS detected that two single nucleotide polymorphisms (SNPs) on BTA7 (ARS-BFGL-NGS-35463 and Hapmap23838-BTA-163815) and one SNP on BTA12 (ARS-BFGL-NGS-2915) significantly affected fat percentage (FAR). The significance of ARS-BFGL-NGS-35463 on BTA7 was confirmed by individual genotyping in all pooled samples. Moreover, association analysis between SNP and FAR in 803 Japanese Black cattle revealed a significant effect of SNP on FAR. Thus, further investigation of these regions is required to identify FAR-associated genes and mutations, which can lead to the development of DNA markers for marker-assisted selection for the genetic improvement of beef quality.
Introduction
Beef marbling is an important trait to determine meat quality in cattle. In Japan, beef marbling is evaluated as Beef Marbling Standard (BMS) number according to the degree of fat deposition in the rib-eye area. BMS is the most influential factor to determine the market price and thus is economically important in the beef industry. Several previous studies have reported BMS-associated genes and mutations in Japanese Black cattle. The mutations in endothelial differentiation sphingolipid G-protein-coupled receptor 1 (EDG1), akirin 2 (AKIRIN2), titin (TTN) and ribosomal protein L27a (RPL27A) have been reported to be associated with beef marbling in Japanese Black cattle (Sasaki et al. 2009; Yamada et al. 2009a,2009b,2009c,2009d). These findings suggested that single nucleotide polymorphisms (SNPs) associated with BMS on these genes would be useful for effective marker-assisted selection to increase marbling levels in Japanese Black cattle. Therefore, the development of additional DNA markers could contribute to the further genetic improvement of beef quality.
BMS number is evaluated by visual inspection, which involves the collation of the actual meat with standard images of each grade, by authorized experts called graders (Shiranita et al. 2000). Several studies have reported that BMS is graded primarily on the basis of fat area ratio to rib eye area (FAR); however, this would also be influenced by the coarseness and fineness of marbling particles and the shape of the rib-eye (Kuchida et al. 1999, 2001; Hasegawa et al. 2004; Hamasaki et al. 2005). In this estimation method, the score may change due to individual differences, and detailed beef traits, such as fineness of marbling and shape of the rib-eye in breeding, are impossible to reflect (Kuchida et al. 2006).
A computer image analysis has been developed to digitalize complex shapes of objects (Takagi & Shimoda 1991), which can be used to obtain more detailed carcass information. Kuchida et al. (2006) have attempted to digitalize the degree of coarseness and fineness of marbling particles and the complexity of rib-eye shape and have estimated genetic parameters for these traits. The findings of this study enabled the objective and in-detail obtainment of beef traits to evaluate quality. In addition to FAR, applying these image analysis traits as additional BMS data for breeding leads to greater improvement in beef quality.
Since high-density SNP genotyping arrays were developed, genome-wide association study (GWAS) has been performed to identify quantitative trait loci (QTL). Although genotyping costs for SNP arrays have gradually decreased, the process remains costly. To reduce the costs of SNP arrays, a DNA pooling method, which genotypes pooled DNA samples to estimate allele frequency differences for each SNP between pooled high and low groups, has been developed. Recently, some pool-based GWASs have been performed to assess economic traits in cattle. Huang et al. (2010) have conducted a pool-based GWAS for fertility traits in Holstein cows. They detected 22 SNPs associated with fertilization rate and five SNPs associated with blastocyst rate. Gambra et al. (2013) have also performed pool-based GWAS for milk proteins using crossbred populations of Holstein × Jersey and have identified 25 SNPs associated with κ-casein concentration and 36 SNPs associated with β-lactoglobulin concentration. These results indicate that pool-based GWAS can be useful to identify QTL and reduce analysis cost.
The objective of this study was to identify genomic regions associated with fat-related traits measured by a computer image analysis using pool-based GWAS in Japanese Black cattle.
Materials and Methods
Animals and traits
We used 1836 Japanese Black cattle from Hyogo prefecture in Japan. They were steers or heifers, slaughtered at 31.9 months of age in average, and graded during 2010 to 2012. Genomic DNA was extracted from each 50 mg Longissimus cervicis muscle sample using standard phenol-chloroform method.
We used four fat-related traits for pooling GWAS: coarseness index of marbling (CIM), fineness index of marbling (FIM), the number of small marbling flecks (NSF) and FAR (Nakahashi et al. 2008). These traits were measured by image analysis. To analyze the traits, high-quality digital images of the carcass cross-section were taken between the sixth and seventh ribs by photographing equipment developed by Kuchida et al. (2001). These were calculated by dividing all pixels of the fat image in the rib-eye area. The rib-eye image was binarized as lean and fat using the image analysis program. The binarized image was thinning with 10 rounds and removing thehairline. The remaining pixels of fat were defined as large fat. CIM is the percentage of large fat pixel in all marbling pixels. FIM is the percentage of the number of small marbling flecks, the size of which is from 0.01 to 0.5 cm2, in the rib-eye area. NSF is the number of small marbling flecks (Nakahashi et al. 2008). The phenotypic data of these traits were available for each animal. We also added BMS data for statistical analysis (Table S1).
DNA pooling construction and pooled DNA genotyping

We selected the highest and lowest 100 individuals of the e-value distribution for each image analysis trait to make two DNA pools (high and low group). In order to avoid overrepresentation of a single sire, the number of progeny from each sire was limited to less than 10 for each group. At the same time, each sire must have at least one offspring in each group. Finally we confirmed that individuals were selected from the highest or lowest 15% of the whole e-value distribution (Table 1).
| Trait | Pool | N | Rank (e-value) | Mean | SD |
|---|---|---|---|---|---|
| CIM | High | 100 | 1–269 (0.1061…0.0248) | 0.0454 | 0.0135 |
| Low | 100 | 1593–1836 (−0.0261…−0.0656) | −0.0406 | 0.0096 | |
| FIM | High | 100 | 1–224 (1.0379…0.3360) | 0.5903 | 0.1721 |
| Low | 100 | 1610–1836 (−0.3382…−0.9114) | −0.5255 | 0.1116 | |
| NSF | High | 100 | 1–271 (79.8626…18.0986) | 36.2960 | 11.2510 |
| Low | 100 | 1574–1836 (−19.0993…−56.2215) | −30.8556 | 7.1930 | |
| FAR | High | 100 | 1–231 (0.0836…0.0331) | 0.0503 | 0.0102 |
| Low | 100 | 1629–1836 (−0.0322…−0.0931) | −0.0537 | 0.0133 |
- Summary statistics include the total number of phenotype records in each pool (N), the rank of residuals (e-value) in 1836 individuals, mean and standard deviation (SD) for each trait. CIM, coarseness index of marbling; FIM, fineness index of marbling; NSF, the number of small marbling flecks; FAR, the fat area ratio to rib-eye area.
Two DNA pools were constructed by taking equivalent amounts of DNA from each selected sample of the high or low group. A final concentration of DNA pools was 50 ng/μL. Pool-based genotyping was performed using Illumina BovineSNP50v2 BeadChip. Genotype calls and normalized intensities from green and red channels were determined by GenomeStudio software (Illumina, Tokyo, Japan).
Estimation of pooled allele frequency and association analysis
We performed pool-based genotyping with three replicate arrays in each pool. The Illumina GenomeStudio genotype module provides the pooled allele frequency estimated as the ‘B-allele frequency’ (Peiffer et al. 2006), and the Illumina method can estimate the accurate allele frequency in pooled samples (Uemoto et al. 2012). These estimated allele frequencies are obtained by interpolation of the known allele frequencies of three canonical genotype clusters represented on a polar coordinate view that originated from an individual genotyping of the Illumina BovineSNP50v2 BeadChip reference sample.
To analyze the effect of SNPs on each trait, we used average B-allele frequencies from the results of three arrays in each pool. All 54 609 SNPs went through quality control that excluded SNPs with minor allele frequencies (MAF) <0.05 in either high or low groups. A total of 27 618 SNPs in CIM, 27 390 SNPs in FIM, 27 457 SNPs in NSF and 27 464 SNPs in FAR on 29 autosomal chromosomes were used for the association analysis.
A Z2-score test was used to determine the significance of single-marker association by testing the null hypothesis H0: 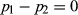 , where p1 and p2 are the allele frequencies in the high and low groups, respectively. The error variance associated with the pooled allele frequency estimates can be split into two components: the sampling error variance Vs and the experimental error variance VE (Sham et al. 2002; Pearson et al. 2007). Because most of the experimental errors in pooling are attributed to a variance in the arrays (Macgregor 2007; Uemoto et al. 2012), we assumed VE as an array variance.
, where p1 and p2 are the allele frequencies in the high and low groups, respectively. The error variance associated with the pooled allele frequency estimates can be split into two components: the sampling error variance Vs and the experimental error variance VE (Sham et al. 2002; Pearson et al. 2007). Because most of the experimental errors in pooling are attributed to a variance in the arrays (Macgregor 2007; Uemoto et al. 2012), we assumed VE as an array variance.



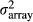 is the array variance, and Nrep is the number of replicate arrays (three replicates). The Z2-statistics are approximately distributed as Chi-square distribution with degree of freedom (df) = 1. Genome-wide significance level was determined by Bonferroni correction. The inflation factor (λ) was also calculated by dividing median of all Z2-statistics by 0.456 (Devlin & Roeder 1999). All analyses were performed with R software (http://www.r-project.org).
is the array variance, and Nrep is the number of replicate arrays (three replicates). The Z2-statistics are approximately distributed as Chi-square distribution with degree of freedom (df) = 1. Genome-wide significance level was determined by Bonferroni correction. The inflation factor (λ) was also calculated by dividing median of all Z2-statistics by 0.456 (Devlin & Roeder 1999). All analyses were performed with R software (http://www.r-project.org).Individual genotyping
The most significant SNP on each chromosome for FAR (ARS-BFGL-NGS-35463 on BTA 7 and ARS-BFGL-NGS-2915 on BTA 12) detected by GWAS were individually genotyped using high and low group animals (n = 200). The genotyping was carried out by PCR-RFLP (restriction fragment length polymorphism). Primers, PCR conditions and restriction enzymes used in PCR-RFLP are shown in Table S3.
Statistical analysis
We verified the effect of the most significant SNP on BTA 7 (ARS-BFGL-NGS-35463) on FAR using 803 individuals, which were randomly selected from 1836 individuals. This population was composed of progeny of sires, each having at least 10 offspring (Table S4).
FAR was analyzed by generalized least squares method implemented in JMP13 (SAS Institute Inc., Cary, NC, USA). The association between genotype and FAR was tested by analysis of variance (ANOVA) and Tukey–Kramer's Honestly Significant Difference (HSD) test. Analytical model included the effect of overall mean, genotype (three levels), slaughter year, slaughter month, sex of animals, linear covariate for inbreeding coefficient, and linear and quadratic covariates for age at slaughter.
Results
Pool-based GWAS
A total of 521 animals were selected for pool-based GWAS. SNP quality was assessed and SNPs with MAF (<0.05) in the high or low group were excluded. A total of 27 618 SNPs in CIM, 27 390 SNPs in FIM, 27 457 SNPs in NSF, and 27 464 SNPs in FAR were used. The obtained B-allele frequencies for each SNP in DNA pools were calculated from the raw signals of red and green channel intensities. The average B-allele frequencies of three replicate arrays were used for statistical analysis.
The difference of the pooled allele frequency between high and low groups was tested to detect SNPs associated with each trait. The λ values of CIM, FIM, NSF and FAR were 1.0, 1.0, 1.0 and 1.2, respectively, and thus the results successfully accounted for population stratification. Bonferroni correction was performed to determine the significant threshold of genome-wide significance at 5% (P-value). P-values were 1.81 × 10−6 and 1.83 × 10−6 for CIM and FIM, respectively, and 1.82 × 10−6 for NSF and FAR. Genome-wide plots of P-values are shown in Figure 1. Two SNPs on BTA7 (ARS-BFGL-NGS-35463 detected on 17.5 Mbp and Hapmap23838-BTA-163815 detected on 28.5 Mbp) and one SNP on BTA12 (ARS-BFGL-NGS-2915 detected on approximately 68.1 Mbp) had significant effects on FAR. These significant SNPs with a genome-wide significance of 5% are listed in Table 2. No significant SNPs were detected for CIM, FIM or NSF at 5% genome-wide significance level.

| SNP name | BTA | Position (bp) (UMD3.1)† | Nearest gene | Distance (bp)‡ | P-value§ |
|---|---|---|---|---|---|
| ARS-BFGL-NGS-35463 | 7 | 17490481 | ARHGEF18 | Within | 8.55 × 10−8 |
| Hapmap23838-BTA-163815 | 7 | 28529552 | LOC100848523 | 16786 | 2.19 × 10−7 |
| ARS-BFGL-NGS-2915 | 12 | 68060288 | LOC101902424 | 16426 | 1.11 × 10−6 |
- †http://www.bovinegenome.org/cgi-bin/gbrowse/bovine_UMD31/. ‡Positive numbers represent distances downstream (bp) from the nearest genes. §The significant threshold of genome-wide significance at 5% accounting for multiple testing by Bonferroni correction was P = 1.82 × 10−6 for FAR. SNP, single nucleotide polymorphism; FAR, the fat area ratio to rib-eye area
Individual genotyping
Two and one significant SNPs on BTA7 and BTA12, respectively, were detected by pool-based GWAS for FAR. To validate pooling methods and confirm significant SNPs detected by pool-based GWAS, the most significant SNPs in respective chromosomes were selected and individually genotyped by PCR-RFLP in all pooled samples (Table 3).
| BTA | SNP | Group | Minor allele frequency | Genotype result (n) | P-value | ||||
|---|---|---|---|---|---|---|---|---|---|
| Pool-based | Individual genotyping | AA | AG | GG | Pool-based | Individual genotyping | |||
| 7 | ARS-BFGL-NGS-35463 | High | 0.402 | 0.435 | 21 | 45 | 34 | 8.55 × 10−8 | 0.000153 |
| Low | 0.159 | 0.255 | 3 | 45 | 52 | ||||
| 12 | ARS-BFGL-NGS-2915 | High | 0.120 | 0.140 | 8 | 12 | 80 | 1.11 × 10−6 | 0.110 |
| Low | 0.319 | 0.200 | 3 | 34 | 63 | ||||
- SNP, single nucleotide polymorphism; FAR, the fat area ratio to rib-eye area.
Difference in allele frequencies by individual genotyping between the high and low groups was tested using Chi-square test. P-values were 0.000153 and 0.110 for ARS-BFGL-NGS-35463 on BTA7 and ARS-BFGL-NGS-2915 on BTA12, respectively. The significance of the effect of SNP on BTA7 was confirmed; therefore, this SNP was used for further analysis to evaluate its effect on FAR. The effect of the most significant SNP on BTA12 detected by individual genotyping was not significant.
Validation analysis
As detected by individual genotyping, ARS-BFGL-NGS-35463 was significantly associated with FAR in the high and low groups. This SNP was genotyped by PCR-RFLP in 803 Japanese Black cattle to confirm the effect of SNP on FAR. ANOVA revealed that the effect of ARS-BFGL-NGS-35463 on FAR was highly significant (P = 0.0044) (Table 4). Tukey–Kramer HSD test was performed to further investigate the effect of ARS-BFGL-NGS-35463 on FAR. The least squares means of FAR among genotypes are summarized in Table 4. HSD test revealed that the AA type exhibited 2.2 percentage points higher FAR than GG type (P < 0.05).
| Trait | AA (n = 75) | AG (n = 344) | GG (n = 384) | P-value |
|---|---|---|---|---|
| FAR (%)† | 0.443 ± 0.0095A | 0.431 ± 0.0079AB | 0.421 ± 0.0075B | 0.0044 |
- A,BMeans with different superscripts differ significantly at P < 0.05 (Tukey–Kramer Honestly Significant Difference test). †Values are least square means ± SE.
Discussion
Image analysis traits used for pool-based GWAS
Pool-based GWAS was performed for CIM, FIM, NSF and FAR, which can lead to a more objective and detailed evaluation of meat quality. These data were collected by computer image analysis. The estimated heritabilities of these traits in this study ranged from 0.37 to 0.59, indicating that these traits were influenced by a sufficient portion of genetic variance in Japanese Black cattle.
BMS number is affected by the fat levels, size, number, shape and valance of marbling distribution. These traits are visually evaluated by qualified graders and may vary due to individual differences (Kuchida et al. 2006). The estimation method by computer image analysis has been established for an objective evaluation of marbling features (Kuchida et al. 1999). In this study, pool-based GWAS was performed for four traits measured by computer image analysis.
For FAR, three significant SNPs were detected and one candidate region was identified on BTA7. Although some reports regarding candidate genes for BMS are available, no responsible genes have been identified. In our population, the heritability of both BMS (0.388) and FAR (0.592) was high (Table S2). Genetic correlation between BMS and FAR was almost unity (0.99), indicating that the identification of responsible genes for FAR would lead to the improvement of BMS. Conversely, no significant SNP was detected for CIM, FIM or NSF. In the present study, the heritabilities of these traits ranged from 0.37 to 0.49 (Table S2). Previous studies have also reported high heritabilities of CIM and FIM, such as 0.47 and 0.43 (Osawa et al. 2008), 0.50 and 0.38 (Nakahashi et al. 2012), and 0.60 and 0.49 (Kato et al. 2014), respectively. However, these traits were not significant because they might be controlled by a large number of SNPs with small effects, whereas FAR may be characterized by an SNP with large effect as indicated by the result of our GWAS. Although no SNPs reached the genome-wide significance threshold (P = 1.81 × 10−6 for CIM, P = 1.83 × 10−6 for FIM, and P = 1.82 × 10−6 for NSF), some SNPs showed relatively low P-values. SNP with the lowest P-values was observed for CIM on BTA8 (P = 4.25 × 10−6), SNP for FIM on BTA15 (P = 4.04 × 10−6), and SNP for NSF on BTA4 (P = 2.46 × 10−6). For NSF, a SNP located on BTA7 also showed as low P-value (P = 2.55 × 10−6) as SNP with the lowest P-value on BTA4, and it was located within the candidate region for FAR. However, no group of SNPs around the SNP on BTA7 showed low P-value for NSF. Furthermore, the genetic correlation between NSF and FAR was low (−0.08) (Table S2). Therefore, we considered that this SNP might be detected by experimental errors. The genomic regions with a group of SNPs with low P-value are likely to be candidate regions for each trait. Further study of these regions is required to identify QTL for these traits.
Fat percentage in rib-eye area
In this study, a cost-effective pooling GWAS was performed. Three FAR-associated SNPs were detected at the significance level of 0.05. Two significant SNPs ranged between 17.5 and 28.5 Mbp on BTA7, and one significant SNP was detected approximately 68 Mbp on BTA12.
To assess the validity of our methods and to confirm the significance of SNPs detected by pool-based GWAS, the most significant SNPs on BTA7 and BTA12 were individually genotyped. For ARS-BFGL-NGS-35463 on BTA7, allele frequencies between the high and low groups were compared. For ARS-BFGL-NGS-35463, difference in individual allele frequencies between the high and low groups was 0.180, whereas that in pool-based allele frequencies was 0.243. For ARS-BFGL-NGS-2915 on BTA12, difference in individual allele frequencies between the high and low groups was 0.060, whereas that detected by pool-based allele frequencies was 0.199. These results indicate that difference in individual allele frequencies between the high and low groups was smaller than that in pool-based allele frequencies for both SNPs. Chi-square test was performed to test the difference in individual allele frequencies between the high and low groups for both SNPs. No significance was detected for ARS-BFGL-NGS-2915 on BTA12. The DNA pooling method could increase the experimental error in allele frequency estimation because of pooling constriction and array variance (Uemoto et al. 2012). The significant SNP on BTA12 might be detected by such experimental errors. For the significant SNP on BTA7 detected by pool-based GWAS, an association analysis between SNP and FAR was performed in Japanese Black cattle, and the result confirmed the significant effect of SNP on FAR (Table 4).
ARS-BFGL-NGS-35463 on BTA7 is located in an intron of the ARHGEF18 (Rho/Rac guanine nucleotide exchange factor 18) gene. ARHGEF18 encodes guanine nucleotide exchange factors, and it directly controls the activation of Rho guanosine triphosphatases. Blomquist et al. (2000) have reported that the ARHGEF18 gene, a member of Rho-growth-hormone releasing factor family, and the overexpression of the ARHGEF18 gene induced the formation of actin stress fibers and stimulated serum response factor-mediated gene transcription in a Rho-dependent manner in humans.
We searched the candidate regions 5 Mbp upstream and downstream of the most significant SNPs on BTA7 for the candidate genes. In a previous study, Watanabe et al. (2008) reported the LD (Linkage Disequilibrium) block size ranged from 22 K to 2.6 Mbp in Japanese Black cattle from a variety of regions in Japan. The Hyogo population has been bred through uniquely closed breeding, which presumably resulted in wider LD block (Honda et al. 2001). Thus, the 5 Mbp would be enough to detect the responsible gene. The 12.5–22.5 Mbp region contained 305 genes, including GCDH (glutaryl-CoA dehydrogenase), ANGPTL4 (angiopoietin like 4), PLIN3 (perilipin 3), PLIN4 (perilipin 4), PLIN5 (perilipin 5) and SIRT6 (sirtuin 6), reported as fat-related genes. GCDH encoded the protein belonging to a member of the acyl-CoA dehydrogenase family. This protein catalyzes the transformation of glutaryl-CoA to crotonyl-CoA by two successive reactions, namely dehydrogenation of glutaryl-CoA to glutaconyl-CoA, and decarboxylation of the glutaconyl-CoA (Hyman & Tanaka 1984). The enzyme encoded by GCDH is involved in carbohydrate metabolism and lipid metabolism. Therefore, GCDH might affect the fat deposition in the beef rib-eye. ANGPTL4 is a member of the angiopoietin-like protein (Angptl) family. The highest expression of this gene was detected in white adipose tissue and the expression was also found in liver, heart, skeletal muscle and intestine. ANGPTL4 expression is controlled under peroxisome-proliferator-activated receptor (PPAR)α in liver, heart, skeletal muscle and intestine, PPARβ/δ in skin, adipose tissue and liver and PPARγ in adipose tissue (Kersten 2005). Thus, this gene is directly involved in regulating lipid metabolism, glucose homeostasis and insulin sensitivity. It suggests that ANGPTL4 might affect FAR in rib-eye. PLIN family (PLIN1, PLIN2, PLIN3, PLIN4 and PLIN5) encodes perilipin known as lipid droplet-associated protein. Perilipin is a modulator of adipocyte lipid metabolism and adipophilins involved in the development and maintenance of adipose tissue (InterPro, http://www.ebi.ac.uk/interpro/). PLIN3, PLIN4 and PLIN5 encode TIP47 (tail-interacting protein of 47 kDa), S3-12, and myocardial lipid droplet protein, respectively. TIP47 is found at the surface of lipid droplets and also within the core and plays a role in the uptake of triglyceride into lipid droplets (Buers et al. 2009). Studies have also reported that when TIP47 expression is suppressed, triglyceride levels in macrophages decrease and when TIP47 levels are increased, the triglyceride content is increased. This indicates that TIP47 encoded by PLIN3 affects fat levels. In subcutaneous adipose tissue and Vastus lateralis, the highest expression among five perilipins was of PLIN4 (Pourteymour et al. 2015). The PLIN4 protein has been reported to be mainly located at the periphery of skeletal muscle fibers, with higher levels in slow-twitch than in fast-twitch skeletal muscle fibers. The rib-eye of cattle is a part of the Longissimus cervicis muscle, and it is also the slow-twitch skeletal muscle. Therefore, it may be proposed that PLIN4 expression in the rib-eye is high, which affects FAR. High expression of PLIN5 can be observed in the brown adipose tissue, skeletal muscle, liver and heart, which have a high oxidative capacity (Wolins et al. 2006). Bosma et al. (2013) have reported that the overexpression of PLIN5 in the skeletal muscle increases the intramyocellular lipid content without impairing insulin sensitivity and increases the expression of genes associated with fatty acid metabolism and oxidation and mitochondrial oxidation. Thus, the overexpression of PLIN5 in the Longissimus cervicis muscle may be associated with FAR in the rib-eye. Sirtuin known as nicotinamide adenine dinucleotide-dependent enzyme is associated with cellular stress resistance, genomic stability, aging and energy homeostasis. Sirtuin family is associated with cellular functions, such as DNA repair, lipid and glucose metabolism, telomeric chromatin maintenance and inflammation (Michan & Sinclair 2007). SIRT6 is a member of the sirtuin family and plays a role in metabolism, inflammation and genome maintenance (Feldman et al. 2013). Xiao et al. (2010) have reported that SIRT6KO mice exhibited symptoms, such as subcutaneous fat loss, slow growth, low blood glucose concentration, hypoglycemia and lymphopenia. Kim et al. (2010) and Schwer et al. (2010) have reported that the liver-specific deletion of SIRT6 and neural-specific deletion of SIRT6 in mice lead to fat gain. Thus, SIRT6 might affect the gain and loss of fat in the beef rib-eye area. All these genes are associated with fat metabolism, and identifying responsible genes and mutations by the further analysis of candidate genes is required.
Some previously reported QTL for fat-related traits on the candidate region were obtained using cattle QTL db. For marbling score and intramuscular fat, three and one QTL, respectively, were reported in this region. For marbling score, Hirano et al. (2007) have performed QTL analysis and identified DIK079-DIK8044 (13.2–26.4 Mbp) in Japanese Black cattle; QTL scan identified DIK2819-UWCA20 (23.4–27.4 Mbp) in commercial American Angus (McClure et al. 2010); and GWAS identified rs41588220-rs43503652 (20.0–21.0 Mbp) and rs43508635-rs43501063 (21.0–22.0 Mbp) in 10 breeds (Saatchi et al. 2014). Peters et al. (2012) performed GWAS for intramuscular fat in Brangus heifers and identified rs41670173-rs109266420 (21.2–21.4 Mbp), rs109286190-rs43706885 (21.5–21.6 Mbp) and rs110410267-rs110835938 (21.5–21.6 Mbp). Considering the QTL overlap in these reports, PLIN3, PLIN4, PLIN5 and SIRT6, located within the region of 20.0–22.0 Mbp, would be probable candidate genes for FAR.
In this study, pool-based GWAS was performed for four fat-related image analysis traits. GWAS revealed two candidate regions for FAR (BTA7 and BTA12). The region identified on BTA7 corresponded with certain reported QTL affecting marbling score and intramuscular fat. Therefore, it is more likely that the responsible genes are located in this region. This information will help to identify novel responsible genes associated with FAR in the rib-eye area in Japanese Black cattle. Further investigation of these regions is required to identify FAR-associated genes and mutations, leading to the development of DNA markers for marker-assisted selection for the genetic improvement of beef quality in Japanese Black cattle.
Acknowledgments
We thank Wagyu Registry Association for providing the pedigree information of Japanese Black. This work was supported in part by JSPS KAKENHI Grant Numbers 16H05015.

