

Using RNA-Seq SNP data to reveal potential causal mutations related to pig production traits and RNA editing
Summary
RNA-Seq technology is widely used in quantitative gene expression studies and identification of non-annotated transcripts. However this technology also can be used for polymorphism detection and RNA editing in transcribed regions in an efficient and cost-effective way. This study used SNP data from an RNA-Seq assay to identify genes and mutations underlying production trait variations in an experimental pig population. The hypothalamic and hepatic transcriptomes of nine extreme animals for growth and fatness from an (Iberian × Landrace) × Landrace backcross were analyzed by RNA-Seq methodology, and SNP calling was conducted. More than 125 000 single nucleotide variants (SNVs) were identified in each tissue, and 78% were considered to be potential SNPs, those SNVs segregating in the context of this study. Potential informative SNPs were detected by considering those showing a homozygous or heterozygous genotype in one extreme group and the alternative genotype in the other group. In this way, 4396 and 1862 informative SNPs were detected in hypothalamus and liver respectively. Out of the 32 SNPs selected for validation, 25 (80%) were confirmed as actual SNPs. Association analyses for growth, fatness and premium cut yields with 19 selected SNPs were carried out, and four potential causal genes (RETSAT, COPA, RNMT and PALMD) were identified. Interestingly, new RNA editing modifications were detected and validated for the NR3C1:g.102797 (ss1985401074) and ACSM2B:g.13374 (ss1985401075) positions and for the COG3:g3.4525 (ss1985401087) modification previously identified across vertebrates, which could lead to phenotypic variation and should be further investigated.
Introduction
The recent availability of massive sequencing technologies provides new tools for the search of causal genes and mutations. Several approaches, such as whole genome sequencing, exome capture and sequencing, chromatin immunoprecipitation sequencing and transcriptome sequencing, have been developed to answer different biological questions (Bai et al. 2012). In particular, RNA sequencing (RNA-Seq) technology is largely used in quantitative gene expression studies as a source of biological information to support the identification of causal mutations underlying the variation of complex traits (Hudson et al. 2012). RNA-seq methodology allows for a comprehensive analysis and quantification of all RNA types expressed in tissues or cells, including mRNA, non-coding RNA and small RNA (Wang et al. 2009). In comparison with gene expression microarrays, RNA-seq technology is able to detect transcripts expressed at low levels and alternative isoforms (Ferraz et al. 2008; Trapnell et al. 2009). During the last few years, the RNA-seq method has also been employed with farm animals and has helped in the selection of candidate genes related to important traits through the comparison of global gene expression profiles between groups of animals that differ in specific traits (i.e. Ramayo-Caldas et al. 2012; Pérez-Montarelo et al. 2014; Wang et al. 2015; Zhang et al. 2015).
Besides allowing the detection of differentially expressed genes, RNA-Seq also enables the identification of previously non-annotated transcripts. Moreover, RNA-Seq technology can be exploited as a method to detect polymorphisms in transcribed regions in an efficient and cost-effective way (Chepelev et al. 2009; Cirulli et al. 2010). In spite of all this, the use of RNA-Seq SNP data to identify candidate genes and mutations in farm animals has not been widely applied (Koltes et al. 2015). To date, only two studies using RNA-Seq for polymorphisms identification in transcribed regions have been reported (Cánovas et al. 2010; Sharma et al. 2012), but further associations and functional interpretation have not been conducted.
Even more, the use of RNA-Seq technology allows the detection of differential allelic expression and post-transcriptional modifications such as RNA editing (Frésard et al. 2015). RNA editing is a posttranscriptional mechanism that generates new transcripts from a limited number of genes in the genome and consists of the chemical alteration of nucleotide bases of RNA molecules (Venø et al. 2012). RNA editing may result in a nucleotide modification, insertion, deletion or substitution in the RNA sequence and may occur in various types of RNA, whether coding or not (Knoop 2011). To the best of our knowledge, only human, mouse, rat, sheep (Shah et al. 2009; Caiment et al. 2010; Danecek et al. 2012; Holmes et al. 2013) and recently chicken (Frésard et al. 2015) studies have identified RNA editing events, except for the known apolipoprotein B (APOB) gene (Greeve et al. 1993).
The main aim of the current study was to use SNP data from a RNA-Seq assay to successfully identify polymorphisms underlying production trait variations in the IBMAP (Iberian × Landrace) experimental porcine population. RNA-editing phenomena were detected from RNA-Seq SNP data and validated for some interesting genes.
Materials and methods
Animal selection, RNA processing and sequencing
The animal material used in the present study was derived from a backcross generated from the IBMAP population. The IBMAP pig population (Iberian × Landrace crosses, including F2, F3 generations and backcrosses) was generated to identify QTL, genes and causal mutations responsible for the variation in production and meat quality traits in pigs, given the remarkable differences existing for such traits between the Iberian and Landrace parental lines (Serra et al. 1998). Results of QTL scans revealed the existence of QTL for growth, fatness and premium cut yields in porcine chromosomes SSC1, SSC2, SSC4, SSC5, SSC6, SSC9, SSC11, SSC13, SSC14, SSC17 and SSCX (Óvilo et al. 2000, 2002; Varona et al. 2002; Mercadé et al. 2005; Fernández et al. 2012, 2014a,b).
The backcross was generated from three Iberian boars mated with 30 Landrace sows (F0) to produce 70 F1 animals. Five F1 boars were mated with 25 Landrace sows, and 187 backcross animals were obtained. All pigs were grown on an experimental farm under standard conditions. Animal manipulations were performed according to the Spanish Policy for Animal Protection RD1201/05, which meets European Union Directive 86/609 concerning the protection of animals used in experimentation. The animals were slaughtered at an approximate age of 175 days. Phenotypic traits related to growth, fatness and premium cut yields were measured in all backcrossed animals as previously described (Fernández et al. 2012) (Table 1).
| Trait description | n | Mean | SD |
|---|---|---|---|
| Body weight at 150 days (kg) | 159 | 79.13 | 10.49 |
| Backfat thickness at 75 kg | 159 | 12.69 | 1.50 |
| Intramuscular fat | 124 | 2.06 | 0.70 |
| Ham weight (kg) | 154 | 10.22 | 1.39 |
| Shoulders weight (kg) | 154 | 5.43 | 0.80 |
| Bone-in-loin weight (kg) | 153 | 7.09 | 1.03 |
The most extreme animals for growth and fatness were selected as described in Pérez-Montarelo et al. (2014). Briefly, a principal components analysis of the backcrossed animals was performed according to four indicators for growth and fatness traits. The 10 male pigs from the same slaughter batch with the most extreme phenotypes, according to the first principal component, were selected for this study and divided into two groups. The five males showing the highest values of growth and fatness indicators were assigned to the High (H) group, and the five males showing the lowest values for these traits to the Low (L) group. The mean values of the four indicators in the H and L groups were respectively 0.92–0.74 kg/day average daily gain, 16.2–11.6 mm of backfat thickness, 12.6–16.7% of C18:2(n − 6) in backfat and 8.1–11.9% of C18:2 (n − 6) in intramuscular fat.
Hypothalamic and hepatic tissue samples from the 10 selected animals were collected at slaughter, immediately frozen in liquid nitrogen and stored at −80 °C until analyzed. Total RNA was extracted using the Ribopure kit (Ambion) to produce high-quality total RNA, following the manufacturer's recommendations, and quantified using a NanoDrop-100 spectrophotometer. The integrity of the RNA was assessed using an Agilent 2100 Bioanalyzer. The RNA integrity value of the samples ranged from 7.1 to 8.1. Paired-end libraries with fragments of 300 bp were prepared using the TruSeq RNA Sample Prep Kit v2 (Illumina Inc.) for each sample. Multiplex sequencing of the libraries was performed on an Illumina Hi-Seq 2000 (Fasteris SA) with three samples per lane at the Centro Nacional de Análisis Genómico, according to the manufacturer's instructions, generating paired-end reads of 75 bp. The raw sequence data have been deposited in the GEO expression database under accession nos. GSE51968 and GSE75850.
RNAseq data analysis
Quality of the raw sequencing data was determined with fastqc (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). trim galore (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) was used to qualitatively trim the data using default settings and to remove the sequencing adaptors and poly A and T tails (stringency of 6 bp), keeping only paired-end reads for which both pairs were longer than 40 bp. One of the 10 samples was discarded for further analyzes due to quality problems (Pérez-Montarelo et al. 2014).
SNP calling
The RNA-seq data filtering, mapping and polymorphisms (single nucleotide variant, SNV) calling was conducted with clc genomics workbench software (www.clcbio.com). Filtered reads were mapped against the pig reference genome (Sscrofa 10.2) based on Mortazavi et al. (2008). Mapping parameters were set at a maximum of two gaps or mismatches per read, and the distance between pairs was set to 50 bp (inner-mean distance) and a standard deviation of 150 bp. Quality-based variant detection from clc genomics workbench was used to perform the SNV detection, with quality parameters set as: minimum quality of central base at 20, minimum quality of surrounding bases (3 pb) at 15, minimum coverage at 10× and a minimum variant frequency at 20%. The quality-based variant detection method in clc genomics workbench is based on the neighborhood quality standard algorithm described by Altshuler et al. (2000) and Brockman et al. (2008), using the PHRED quality score surrounding the potential SNP. The SNP calling was conducted within tissue, using all reads from the same tissue for polymorphism identification. After, individual genotypes were identified for each single variant. Those SNVs segregating in the sequenced animals, heterozygous or alternative homozygous among the sequenced animals, were considered actual single nucleotide polymorphisms. SNPs showing divergent genotypes between the H and L groups (homozygous or heterozygous in one group and alternative allele homozygous in the other group, and considering absence of genotype equal to the reference allele at this position) were selected as potential informative SNPs.
Variant annotation, functional enrichment and effect predictions
The Ensembl Variation database in Biomart (www.ensembl.org/biomart) was used to annotate previously described porcine variants. Functional annotation and enrichment analysis were conducted using the fatigo tool from babelomics (Medina et al. 2010) and human genome annotation as a reference. Adjusted P-values, based on the false discovery rate (FDR) method of accounting for multiple testing (Benjamini & Hochberg 1995), were used to identify Gene Ontology enrichment terms. The predictprotein (www.predictprotein.org) tool was used to predict amino acid change effects on protein structure and function, and regrna (http://regrna.mbc.nctu.edu.tw/) was used to identify changes in regulatory motifs.
SNP validation
A set of 32 SNPs were selected for validation based on functional and positional criteria and potential effect relevance. Validation was conducted by standard Sanger sequencing on cDNA synthesized from RNA from both tissues from the same animals used for the RNA-Seq assay. Primer pairs were designed from exon to exon to avoid genomic DNA amplification (Table S1). The PCR reactions were performed in a final volume of 25 μl, containing 2.5 μl of cDNA, 1 unit of Taq polymerase (Biotools), specific buffer, 2.5 mm of dNTPs and 0.5 μm of each primer. Thermocycling was carried out under the following conditions: 94 °C for 5 min, 40 cycles of 94 °C for 45 s, 60 °C for 45 s and 72 °C for 45 s, with a final extension of 72 °C for 10 min. The PCR reactions were carried out in a GeneAmp PCR System 9700 (Applied Biosystems). The PCR products were purified with the GFXTM PCR DNA purification kit (GE Healthcare), according to the manufacturer's protocol. PCR products were sequenced with both forward and reverse primers using the 3100 BigDye® Terminator v3.1 Matrix Standard in a 3730 DNA Analyzer (Applied Biosystems).
SNP genotyping and association analyses
Fourteen candidate genes – retinol saturase (RETSAT); megakaryoblastic leukemia (translocation) 1 (MKL1); serpin peptidase inhibitor, clade E (SERPINE1); synoviolin 1 (SYVN1); leucyl-tRNA synthetase (LARS); coatomer protein complex subunit alpha (COPA); cell adhesion molecule 3 (CADM3); ephrin-A1 (EFNA1); palmdelphin (PALMD); calponin 3 (CNN3); rhomboid like 2 (RHBDL2); janus kinase 1 (JAK1); hydroxyacid oxidase (glycolate oxidase) 1 (HAO1); and RNA (guanine-7-) methyltransferase (RNMT) – carrying SNPs identified in the RNA-Seq assay and subsequently validated, were selected for association analyses due to their potential role in biological processes involved in traits related to growth, fatness and premium cut yields (functional candidate genes) or their putative positions within QTL intervals (positional candidate genes).
Functional candidate genes
Polymorphisms located in seven functional candidate genes (described below) were selected due to their functional relationship with the analyzed traits. The RETSAT gene encodes a retinol saturase, which promotes adipogenesis and is downregulated in obesity (Schupp et al. 2009). The SERPINE1 gene encodes a serpin peptidase inhibitor that has been associated with muscling, growth, fat accretion and meat quality in pigs (Weisz et al. 2012). The MKL1 gene, also known as MRTF-A, encodes the megakaryoblastic leukemia protein, which interacts with the transcription factor myocardin and which is a key regulator of muscle cell differentiation and remodeling (Mokalled et al. 2012). The HAO1 gene encodes a hydroxyacid oxidase, which constitutes a liver-specific peroxisomal enzyme that oxidizes glycolate to glyoxylate and is a known diabetes marker (Recalcati et al. 2003). The RNMT gene (similar to LOC100626038) encodes a methyltransferase involved in mRNA processing, stability and translation required for cell proliferation (Aregger & Cowling 2013). The RHBDL2 gene encodes an integral membrane protein that releases soluble growth factors by proteolytic cleavage of certain membrane-bound substrates (Pascall & Brown 2004); specifically it releases epidermal growth factor, which mediates adipocyte differentiation (Harrington et al. 2007). The SYVN1 gene encodes a synovial apoptosis inhibitor that removes unfolded proteins; it is implicated together with PGC-1β in the regulation of mitochondrion number, respiration and basal energy expenditure in adipose tissue, and therefore, it determines weight and accumulation of white adipose tissue (Fujita et al. 2015).
Ten SNPs in these seven candidate genes were genotyped: RETSAT:g.3320A>C, RETSAT:g.3453A>C, RETSAT:g.3661indelT, RETSAT:g.3962A>G, SERPINE1:g.1249A>G, MKL1:g.10685T>C; HAO1:g.7569A>T, RNMT:g.185A>C, RHBDL2:g.19048A>G and SYVN1:g.2895G>T (submitted to SNPdb as ss1985400207, ss1985400206, ss1985400205, ss1985400204, ss1985400218, ss1985400213, ss1985400216, ss1985400217, ss1985400214, ss1985400201 respectively).
Positional candidate genes
Polymorphisms located in seven genes mapped within QTL intervals for the analyzed traits were selected: the LARS gene maps to 153 Mb on SSC2, close to the QTL for premium cut yield identified previously (Fernández et al. 2012). This gene encodes a leucyl-tRNA synthetase, which catalyzes the ATP-dependent ligation of l-leucine to tRNA(Leu), responsible for specific hepatopathies (Casey et al. 2012); the JAK1 gene maps to 135 Mb on SSC6, within the QTL interval for backfat thickness and premium cut yields previously identified (Óvilo et al. 2000; Varona et al. 2002; Fernández et al. 2012) and encodes a protein-tyrosine kinase, directly implicated in insulin resistance in obese individuals (Khan et al. 2015). Two missense SNPs in the LARS gene (g.33280A>C and g.7010G>A) and two nonsense SNPs in the JAK1 gene (g.18273A>G and g.12288A>G) were selected for genotyping.
Five other functional gene candidates underlying the QTL effects on SSC4 for fatness and premium cut yields were also analyzed: PALMD:g.210G>C, CNN3:g.17250A>G, COPA:g.48255C>T, CAMD3:g.21844T>C and EFNA1:g.5633T>C (submitted to SNPdb as ss1985400203, ss1985401075, ss1985400207, ss1985400208 and ss1985401076 respectively). The palmdelphin (PALMD) gene maps to 129 Mb and encodes a protein involved in p53 phosphorylation (Dashzeveg et al. 2014), which is crucially involved in the development of insulin resistance through the modulation of cell apoptosis (Minamino et al. 2009); the calponin 3 (CNN3) gene maps to 134 Mb and encodes a protein associated with growth traits in pig (Tang et al. 2014); the coatomer protein complex subunit alpha (COPA) gene maps to 98 Mb and encodes a coat protein responsible for the transport between the endoplasmic reticulum and Golgi compartments, which may play an important role in muscle development (Qiu et al. 2010); the ephrin-A1 (EFNA1) gene maps to 103 Mb and encodes a protein-tyrosine kinase receptor, which is implicated in angiogenesis (Deroanne et al. 2003) and consequently in adipose tissue expansion (Rupnick et al. 2002).
The RETSAT (ss1985400204, ss1985400205, ss1985400206, ss1985400207), MKL1 (ss1985400213) and SERPINE1 (ss1985400218) SNPs were genotyped in 122 backcrossed animals with available samples and registered phenotype using Sanger sequencing. Primer pairs are described in Table S1. Additionally, (SYVN1) (ss1985400201), LARS (ss1985400202, ss1985400203), COPA (ss1985400208), CADM3 (ss1985401078), EFNA1 (ss1985400210), PALMD (ss1985400211), CNN3 (ss1985400212), RHBDL2 (ss1985400214), JAK1 (ss1985401084, ss1985400215), HAO1 (ss1985400216) and RNMT (ss1985400217) polymorphisms were genotyped using the OpenArray platform at Servei Veterinari de Genètica Molecular (Universidad Autónoma de Barcelona, Spain). Haplotypes were built using phase version 2.1 program (Stephens et al. 2001).

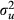 , A being the numerator relationship matrix); and eijk is the random residual term. Dominance effect was not included in the model because it was not significant. Strict Bonferroni correction (α/SNP number) was applied to correct for multiple tests (0.05/19 = 2.6 × 10−3).
, A being the numerator relationship matrix); and eijk is the random residual term. Dominance effect was not included in the model because it was not significant. Strict Bonferroni correction (α/SNP number) was applied to correct for multiple tests (0.05/19 = 2.6 × 10−3).Gene expression analyses
Differential gene expression conditional on SNP genotypes that showed significant associations with production traits were evaluated here using the RNA-Seq expression data (Fernández et al. 2014a,b; Pérez-Montarelo et al. 2014). Transcripts were assembled and quantified in RPKM (reads per kilobase of transcript per million mapped) by clc genomics workbench.
Validation of gene expression differences for RETSAT and PALMD on hepatic tissue were conducted by quantitative PCR (qPCR) in a larger sample set; hepatic samples were from 31 males from the same slaughter batch. The cDNA synthesis was performed using Superscript II enzyme (Invitrogen). Relative transcript quantification was performed on 384 plates using the LightCycler®480 Real-Time PCR System (Roche Diagnostic). Real-time qPCR reactions were performed in a total volume of 20 μl containing 2.5 μl of cDNA (1/20 dilution), 10 μl of Roche LightCycler mix and 0.5 μm of primer pairs (Table S1). All points and samples were run in triplicate as technical replicates, and dissociation curves were analyzed for each individual replicate. Actin, beta (ACTB) and TATA-box binding protein (TBP) were used as control genes.
Gene expression data were normalized by the endogenous genes using genorm software (http:/medgen.ugent.be/~jvdesomp/genorm) and analyzed using a general linear mixed model including the additive effect of the SNP or haplotypes and a random litter effect accounting for the correlations among the records of full sibs.
RNA editing validation
A subset of three SNPs showing different genotypes conditional on the analyzed tissue (hypothalamus or liver) was also validated on genomic DNA (gDNA). Primer pairs were designed (Table S1), and amplification and sequencing were conducted as mentioned above. Furthermore, pyrosequencing protocols (Table S1) were designed to validate the genotype differences between DNA sources (gDNA or cDNA). In addition, gDNA and hypothalamic RNA samples from three pure Iberian and three Iberian × Large White pigs were also added to the validation step.
Results
SNP detection
A total of 839 and 877 millions of paired reads were obtained from hypothalamus and liver RNA-Seq data respectively. After filtering by quality and mapping against the reference genome Sscrofa 10.2, 66% and 77% of the reads matched the reference sequence respectively, and 50% corresponded to annotated genes.
The variant detection analysis revealed a total of 125 488 SNVs in hypothalamus and 125 163 SNVs in liver (Table 2), and 45% of the SNVs were identified in both tissues, giving a total of 192 143 SNVs that were detected against the reference genome in this study. Those SNVs segregating in the sequenced animals were considered as actual SNPs: 97 427 SNPS identified in hypothalamus and 99 290 SNPs in liver. Approximately 65–70% of these SNPs were located in exonic regions already annotated, and roughly 10% of them corresponded to missense SNPs (Table 2). A total of 4396 informative SNPs were detected in hypothalamus and 1862 in liver, including 331 SNPs that were detected in both tissues. Informative SNPs were considered those showing a homozygous or heterozygous genotype in one group and the alternative genotype in the other group.
| Gene | Exon | Coding region | Missense | n > 21 | |
|---|---|---|---|---|---|
| Hypothalamus | |||||
| SNVs | 125 488 | 84 941 | 30 472 | 9588 | 98 869 |
| SNPs2 | 97 427 | 63 252 | 24 527 | 7310 | 77 548 |
| Potential informative SNPs3 | 4396 | 3277 | 835 | 242 | 4396 |
| Liver | |||||
| SNVs | 125 163 | 93 560 | 23 605 | 7707 | 104 621 |
| SNPs2 | 99 290 | 72 792 | 19 092 | 5994 | 76 327 |
| Potential informative SNPs3 | 1862 | 1341 | 383 | 132 | 1862 |
- 1SNPs identified in more than two of the sequenced animals.
- 2SNVs segregating, at least two animals heterozygous or homozygous for the alternative allele, were considered SNPs.
- 3Those SNPs for which the animals were homozygous or heterozygous in one group and alternative allele homozygous in the other group were considered potential informative SNPs.
The enrichment of biological processes affected by the genes that contained the identified SNPs in both tissues (Table 3) revealed that the same general terms – organic acid carboxylic acid, cellular amino acid and derivative and lipid metabolisms – were enriched in both tissues. However, the regulation of the developmental process term was specifically enriched in hypothalamus, in accordance with the fact that it is the main tissue implicated in hormonal coordination, complex patterns of neuroendocrine outputs and homeostatic mechanisms during development (Swaab et al. 2001).
| Gene Ontology terms | No. of genes | Adjusted P-values1 |
|---|---|---|
| Hypothalamus | ||
| Organic acid metabolic process | 80 | 3.69 × 10−4 |
| Regulation of developmental process | 110 | 3.96 × 10−4 |
| Carboxylic acid metabolic process | 78 | 6.29 × 10−4 |
| Celular amino acid and derivative metabolic process | 41 | 6.82 × 10−4 |
| Lipid metabolic process | 105 | 9.07 × 10−4 |
| Liver | ||
| Organic acid metabolic process | 57 | 4.64 × 10−6 |
| Carboxylic acid metabolic process | 57 | 4.64 × 10−6 |
| Lipid metabolic process | 72 | 4.64 × 10−6 |
| Celular lipid metabolic process | 62 | 6.44 × 10−6 |
| Amine metabolic process | 41 | 3.04 × 10−4 |
- 1Adjusted P-values based on false discovery rate method of accounting for multiple testing (Benjamini & Hochberg 1995).
The distribution of the detected SNPs along the porcine chromosomes is shown in Fig. 1. The proportions of SNPs per chromosome were compared with those obtained from the Ensembl Variation Database (Ensembl Variation 82; Sus scrofa Short Variants 10.2). Both distributions were similar (Fig. 1a), but differences were observed when focusing on the potential informative SNPs (P-value = 10−4), especially for some chromosomes such as SSC6, where the number of SNPs was much higher than in the annotated results in the Ensembl database (Fig. 1b).

A set of 32 SNPs (Table 4) was selected for validation. These SNPs met one or more of the following criteria: involvement in biological processes related to the analyzed traits, inducing amino acid changes, location on QTL regions previously described (Óvilo et al. 2000; Varona et al. 2002; Fernández et al. 2012, 2014a,b) and genotype differences between tissues. The sequencing of cDNA from the same hypothalamic and hepatic samples confirmed that 25 out of the 32 detected SNPs were actual SNPs, representing 80% of the total number of tested SNPs. From the remaining SNPs, six were false positives and one of them could not be tested due to unsuccessful amplification (Table 4).
| SNP identification | Chromosome position | SNPdb no. | Gene name | Amino acid change | Validation by Sanger sequencing |
|---|---|---|---|---|---|
| ENSSSCG00000027057g.2895G>T | 2:6195341 | ss1985400201 | SYVN1 | Yes | |
| ENSSSCG00000014401g.102797T>C | 2:151055783 | ss1985401074 | NR3C1 | Yes | |
| ENSSSCG00000014411g.33280A>C | 2:153858779 | ss1985400202 | LARS | Lys614Thr | Yes |
| ENSSSCG00000014411g.7010G>A | 2:153885049 | ss1985400203 | LARS | Asp127Asn | Yes |
| ENSSSCG00000007858g.13374T>A | 3:26152993 | ss1985401075 | ACSM2B | Ser272Thr | Yes |
| ENSSSCG00000008237g.3320A>C | 3:62319781 | ss1985400207 | RETSAT | Lys173Thr | Yes |
| ENSSSCG00000006386g.48255C>T | 4:98081626 | ss1985400208 | COPA | Yes | |
| ENSSSCG00000006386g.48429G>T | 4:98081800 | ss1985401076 | COPA | Yes | |
| ENSSSCG00000006386g.48430G>T | 4:98081801 | ss1985401077 | COPA | Yes | |
| ENSSSCG00000006415g.21844T>C | 4:99236075 | ss1985401078 | CADM3 | Yes | |
| ENSSSCG00000006530g.5633T>C | 4:103449624 | ss1985400210 | EFNA1 | Yes | |
| ENSSSCG00000006582g.1877C>T | 4:104969868 | ss1985401079 | S100A14 | Yes | |
| ENSSSCG00000006727g.49406C>G | 4:112791607 | ss1985401080 | WDR3 | Yes | |
| ENSSSCG00000006874g.210G>C | 4:129990914 | ss1985400211 | PALMD | Yes | |
| ENSSSCG00000006887g.17250A>G | 4:134252015 | ss1985400212 | CNN3 | Yes | |
| ENSSSCG00000000075g.10685T>C | 5:5200373 | ss1985400213 | MKL1 | Val245Ala | Yes |
| ENSSSCG00000028272g.48226C>T | 5:16572785 | ss1985401081 | FP565371.2 | Ala109Val | Yes |
| ENSSSCG00000030076g.27047A>G | 5:69870959 | ss1985401082 | SLC6A13 | No | |
| ENSSSCG00000003247g.4656C>T | 6:52789410 | ss1985401083 | Non-annotated | No | |
| ENSSSCG00000003651g.19048A>G | 6:87893864 | ss1985400214 | RHBDL2 | Met19Val | Yes |
| ENSSSCG00000003809g.18273A>G | 6:135899621 | ss1985401084 | JAK1 | Yes | |
| ENSSSCG00000003809g.12288A>G | 6:135905604 | ss1985400215 | JAK1 | Yes | |
| ENSSSCG00000023489g.4832C>A | 8:75801501 | ss1985401085 | CXCL9 | No | |
| ENSSSCG00000010884g.14128A>G | 10:21630979 | ss1985401086 | Non-annotated | No1 | |
| ENSSSCG00000027815g.4525A>G | 11:22178068 | ss1985401087 | COG3 | Ile83Val | Yes |
| ENSSSCG00000017383g.4388C>A | 12:20231782 | ss1985401088 | AOC3 | No | |
| ENSSSCG00000010487g.9565G>A | 14:116122358 | ss1985401089 | Non-annotated | No | |
| ENSSSCG00000010488g.39756G>A | 14:116244344 | ss1985401090 | Non-annotated | No | |
| ENSSSCG00000010739g.20840C>G | 14:145893720 | ss1985401091 | CTBP2 | Yes | |
| ENSSSCG00000027439g.7569A>T | 17:18863266 | ss1985400216 | HAO1 | Yes | |
| ENSSSCG00000025855g.185A>C | GL893425.2:41788 | ss1985400217 | RNMT | Glu62Ala | Yes |
| ENSSSCG00000025698g.1249A>G | GL894574.1:17487 | ss1985400218 | SERPINE1 | Ile150Val | Yes |
- 1No amplification.
Association studies
The 19 SNPs located in the 14 candidate genes were successfully genotyped in the 122 backcrossed animals and showed minor allele frequencies (MAFs) ranging from 0.08 for SERPINE1:g.1249A>G to 0.46 for COPA:g.48255C>T. Most of the SNPs showed intermediate frequencies, and only four showed a MAF < 0.25 (Table 5). The two SNPs in the JAK1 gene (g.18273A>G and g.12288A>G) appeared fully linked with a MAF = 0.38.
| Polymorphism | SNPdb no. | MAF | Additive effect on the trait (standard error) | |||
|---|---|---|---|---|---|---|
| BFT75 (mm) | HW (kg) | SW (kg) | BLW (kg) | |||
| SYVN1:g.2895G>T | ss1985400201 | 0.10 | 0.398 (0.329) | −0.160 (0.145) | −0.077 (0.079) | −0.164 (0.137) |
| LARS:g.33280A>C | ss1985400202 | 0.38 | 0.304 (0.201) | 0.074 (0.094) | 0.032 (0.051) | −0.013 (0.087) |
| LARS:g.7010G>A | ss1985400203 | 0.38 | 0.304 (0.199) | 0.071 (0.093) | 0.033 (0.051) | −0.010 (0.086) |
| RETSAT:g.3320A>C | ss1985400207 | 0.32 | −0.298 (0.191)* | 0.214 (0.087)* | 0.032 (0.049) | 0.149 (0.081)¥ |
| RETSAT:g.3453A>C | ss1985400206 | 0.29 | −0.322 (0.278) | −0.139 (0.132) | −0.017 (0.072) | 0.011 (0.122) |
| RETSAT:g.3661delinsT | ss1985400205 | 0.14 | 0.042 (0.294) | 0.281 (0.132)* | 0.015 (0.073) | 0.238 (0.121)¥ |
| RETSAT:g.3962A>G | ss1985400204 | 0.45 | −0.063 (0.259) | 0.142 (0.119) | −0.015 (0.064) | 0.118 (0.109) |
| COPA:g.48255 C>T | ss1985400208 | 0.46 | 0.528 (0.180)**€ | −0.018 (0.086) | −0.060 (0.046) | −0.049 (0.078) |
| CADM3:g.21844T>C | ss1985401078 | 0.14 | 0.525 (0.282)¥ | −0.131 (0.140) | −0.109 (0.074) | −0.075 (0.127) |
| EFNA1:g.5633T>C | ss1985400210 | 0.31 | −0.318 (0.194) | 0.021 (0.097) | 0.046 (0.052) | −0.068 (0.087) |
| PALMD:g.210G>C | ss1985400211 | 0.31 | −0.825 (0.200)***€ | 0.112 (0.098) | 0.084 (0.052) | 0.076 (0.092) |
| CNN3:g.17250A>G | ss1985400212 | 0.30 | 0.400 (0.205)¥ | −0.037 (0.096) | −0.051 (0.052) | −0.052 (0.089) |
| MKL1:g.10685T>C | ss1985400213 | 0.40 | 0.034 (0.179) | −0.043 (0.082) | −0.056 (0.044) | −0.023 (0.076) |
| RHBDL2:g.19048A>G | ss1985400214 | 0.32 | −0.187 (0.201) | 0.167 (0.099) | 0.090 (0.051)¥ | 0.164 (0.088)¥ |
| JAK1:g.12288A>G | ss1985400215 | 0.38 | 0.265 (0.213) | −0.020 (0.103) | 0.041 (0.055) | −0.089 (0.093) |
| HAO1:g.7569A>T | ss1985400216 | 0.34 | 0.156 (0.265) | 0.052 (0.122) | 0.045 (0.066) | −0.016 (0.112) |
| RNMT:g.185A>C | ss1985400217 | 0.34 | −0.123 (0.220) | 0.205 (0.099)* | 0.104 (0.050)* | 0.218 (0.087)* |
| SERPINE1:g.1249A>G | ss1985400218 | 0.08 | −0.024 (0.354) | −0.172 (0.163) | −0.045 (0.084) | 0.060 (0.147) |
- BFT75, body weight at 150 days (kg); HW, ham weight; SW, shoulder weight; BLW, bone-in-loin weight.
- ¥P < 0.10; *P < 0.05; **P < 0.005; ***P < 0.0005; €Significant after Bonferroni correction.
The association analysis results (Table 5) revealed coherent effects of the RETSAT polymorphisms (ss1985400207 and ss1985400205) on ham weight, although the association was not significant after multiple testing correction. Both RETSAT:g.3320C and g.3661delT alleles showed similar effects: an increase of around 200 g on mean ham weight. Haplotypes were built for these two polymorphisms (H1: g.3320C/g.3661insT; H2: g.3320C/g.3661delT; H3: g.3320A/g.3661insT), and the association analysis revealed more relevant additive effects than did the single analysis (Fig. 2). A comparison of the H1 (CT) and H2 (C–) haplotypes with H3, showed an increase in ham and bone-in-loin mean weights and leading to a potential reduction in backfat thickness. Effects on premium cut yields were also found for the RNMT polymorphism (ss1985400217): the RNMT:g.185C allele was associated with a 200 g increase in ham weight, a 100 g increase in shoulder weight and a 200 g increase in the bone-in-loin mean weight. Moreover, large effects were detected for the COPA and PALMD polymorphisms (ss1985400208 and ss1985400211) on backfat thickness at 75 kg (Table 5). The COPA:g.48255T allele was associated with an increase in fat deposition of about 0.5 mm, whereas the PALMD:g.210C allele was associated with an increase fat deposition of about 0.8 mm. No effect could be detected for body weight or intramuscular fat.

Additionally, differential gene expression conditional on genotypes for the significantly associated SNPs was tested on the available RNA-Seq data. Expression differences in liver were found for the RETSAT and PALMD transcripts conditional on the respective genotypes (Table 6). Moreover, relative gene expression was measured by qPCR on hepatic samples carrying different RETSAT genotypes:g.3320A>C, g.3453A>C, g.3661delinsT, g.3962A>G (ss1985400207, ss1985400206, ss1985400205 and ss1985400204 respectively). RETSAT:g.3962A>G showed significant expression differences: the g.3962G carriers expressed 0.147 times more than did the A carriers (P-value = 0.02). Moreover, RETSAT haplotypes were also built, and an association analysis with gene expression data was conducted. Four haplotypes were identified in the 31 analyzed samples, AATA (freq = 0.27), CATA (freq = 0.26), CA–T (freq = 0.15) and CCTG (freq = 0.32). The results allowed us to identify RETSAT expression differences between the CCTG and CATA haplotypes (fold change = 0.18, P-value = 0.01). However, PALMD gene expression differences conditional on its genotype could not be validated by qPCR in a larger sample set.
| Transcript | Liver | Hypothalamus) |
|---|---|---|
| RPKM (SD) | RPKM (SD | |
| RETSAT:g.3320C>A; g.3661indelT (ss1985400207; ss1985400205) | ||
| H3 | 85.37 (25.03) | 23.08 (3.74) |
| H1/H2 | 30.17 (4.42) | 27.44 (0.95) |
| Ratio | 2.83 | 0.84 |
| P-value | 0.03 | 0.12 |
| RNMT:g.185A>C (ss1985400217) | ||
| AC | 4.65 (0.27) | 16.55 (1.40) |
| CC | 4.50 (0.42) | 13.58 (3.87) |
| Ratio | 1.03 | 1.21 |
| P-value | 0.59 | 0.28 |
| COPA:g.48255 C>T (ss1985400208) | ||
| CC | 31.29 (0.94) | 49.29 (4.32) |
| TT | 50.19 (10.36) | 45.72 (4.04) |
| Ratio | 0.62 | 1.08 |
| P-value | 0.12 | 0.35 |
| PALMD:g.210G>C (ss1985400211) | ||
| CC | 10.76 (0.21) | 6.39 (1.31) |
| GC | 2.61(2.01) | 5.55 (1.59) |
| Ratio | 4.12 | 1.15 |
| P-value | 0.002 | 0.48 |
- RPKM, reads per kilobase of transcript per million mapped.
RNA editing
Three of the validated SNPs on the nuclear receptor subfamily 3 group C member 1 (NR3C1, ENSSSCG00000014401:g.102797T>C, ss1985401074), component of oligomeric golgi complex 3 (COG3, ENSSSCG00000027815:g.4525A>G, ss1985401087) and Acyl-CoA synthetase medium-chain family member 2B (ACSM2B, ENSSSCG00000007858:g.13374T>A, ss1985401075) revealed different genotypes conditional on the type of analyzed tissue (Table 7), which might indicate tissue-specific allelic expression. The results of validation through Sanger sequencing on genomic DNA showed unexpected genotypes (Table 7 and Fig. 3). Some transcripts showed alleles that are not present in the corresponding gDNA sequence. These results were also validated by genotyping through the pyrosequencing method (Fig. 3). The results as a whole suggest the detection of RNA editing phenomena, i.e. RNA nucleotide substitutions (A>G, A>U, U>A), leading to differences between the final RNA sequence and the DNA region from which it was transcribed.
| COG3:g.4525A>G (ss1985401087) | ACSM2B:g.13374A>T (ss1985401075) | NR3C1:g.102797T>C (ss1985401074) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Hypothalamus | Liver | gDNA | Hypothalamus | Liver | gDNA | Hypothalamus | Liver | gDNA | |
| H-group | |||||||||
| Individual 1 | AG | GG | AA | AT | AA | AA | CT | CT | TT |
| Individual 2 | AG | GG | AA | AT | AA | AA | CC | CC | CC |
| Individual 3 | AG | GG | AA | AA | AA | AA | CC | CC | CC |
| Individual 4 | AG | GG | AA | AT | AA | AA | CC | CC | CC |
| L-group | |||||||||
| Individual 5 | AG | GG | AA | AT | TT | AA | CT | CT | TT |
| Individual 6 | AG | GG | AA | AT | AT | AA | CC | CC | CC |
| Individual 7 | AG | GG | AA | AA | AT | AA | CC | CC | CC |
| Individual 8 | AG | GG | AA | AA | AT | AA | CC | CT | TT |
| Individual 9 | AG | GG | AA | AA | AT | AA | CT | CT | TT |
| Additional sample set | |||||||||
| IB1 | AG | – | AA | AT | – | AA | CC | – | CC |
| IB2 | AG | – | AA | AA | – | AA | CT | – | TT |
| IB3 | AG | – | AA | AT | – | AA | CC | – | CC |
| IB × LW1 | AG | – | AA | AA | – | AA | CC | – | CC |
| IB × LW2 | AG | – | AA | AA | – | AA | CC | – | CC |
| IB × LW3 | AG | – | AA | AT | – | AT | CC | – | CC |

Moreover, genotyping by sequencing was conducted on an additional set of hypothalamic cDNA and gDNA samples from six animals (pure Iberian and Iberian × Large White). The sequencing of these additional animals with different genetic origins revealed the same genotype profiles as previously detected for the three SNPs (Table 7).
Discussion
SNP detection
In the current study, an effort was made to evaluate the usefulness of RNA-Seq technology to successfully identify valuable SNPs. Moreover, detection of powerful candidate genes and mutations underlying production trait variations in the IBMAP population and RNA editing phenomena were detected and validated.
More than 125 000 SNVs were identified in each tissue, and 78% were considered potential SNPs. A SNV was considered as segregating if at least two animals were heterozygous or homozygous for the alternative allele within the analyzed samples. Thus, 22% of the identified SNVs could correspond either to fixed variants in the population studied (Iberian and Landrace vs. Duroc) or errors in the reference sequence (Duroc). Although the assay was focused on gene expression analysis, 35% of the identified SNPs mapped to within intronic regions, which probably points to the existence and sequencing of immature RNAs and a large number of non-annotated transcripts due to the incompleteness of the available porcine genome annotation. Among the SNPs falling within annotated exons, the ratio of synonymous to non-synonymous polymorphisms was 2.2:1 in hypothalamus and 2.4:1 in liver, showing as expected (Schattner & Diekhans 2006) that a large proportion of the identified SNPs do not lead to amino acid changes.
Further analyses were conducted to identify potentially informative SNPs in the H and L groups of the experimental backcross that could lead to causal genes and mutations for growth, fatness and meat yield. Because of the limited number of samples used in the RNA-Seq assay, a strict criterion, SNPs showing a homozygous or heterozygous genotype in one group and the alternative genotype in the other group, was set up. Due to the nature of the backcrossed population, we did not expect to find informative and causative mutations displaying alternative alleles between the established groups. As could be expected, the number of potentially informative SNPs was greatly reduced to 4396 in hypothalamus and 1862 in liver.
The reliability of the sequencing data, read filtering, mapping and SNP calling processes was evaluated using a set of 32 SNPs selected using standard Sanger sequencing. Only five false positive SNPs were detected. A deeper examination of the sequences covering these false positive SNPs allowed us to determine that the main factor influencing this result was likely mapping errors due to the excess of coverage in highly expressed transcripts. The SNP calling from RNA-Seq data, in contrast with whole genome sequence analysis, has to allow for high coverage because it is conditional on gene expression level, which may increase the chance of sequence errors at these highly sequenced genes. Here, the sequencing errors caused by the sequencing technology should also be taken into account, as Illumina technology shows error rates ranging from 0.3% at the beginning of reads to 3.8% at the end of reads (Dohm et al. 2008). Therefore, a more restricted criterion in the SNPs surrounding bases (i.e. from 5 to 10 bp) and an increase in the minimum read per allele (i.e. from 20% to 30%) could reduce the FDR.
The SNP proportions per chromosome identified here do not differ from those of the Ensembl porcine SNP database; however, when focusing on potential informative SNPs, the proportions do differ, especially for SSC6. SSC6 is particularly relevant in the IBMAP population, as a major QTL for growth, fat deposition and conformation has been mapped there (Óvilo et al. 2000; Varona et al. 2002), therefore a large degree of polymorphism is expected in this region between the parental breeds (Iberian and Landrace). Moreover, the LEPR gene is the main candidate for this QTL, as previously reported in several studies (Óvilo et al. 2000, 2002, 2010; Galve et al. 2012), which seems to have a specific behavior and sequence variation in Iberian pigs that is not found in other porcine breeds (Torres-Rovira et al. 2012; Pérez-Montarelo et al. 2015).
Association analyses
Genotyping and association analyses in the backcrossed population were conducted for 19 of the validated SNPs. The frequencies obtained indicate that the selection was successful for the identification of potential informative SNPs, as most SNPs showed intermediate frequencies in the population (MAF > 0.25), which are optimal in association analyses (Tabangin et al. 2009). Here, the limitation in the population size, which makes it difficult to estimate highly significant allelic or genotype effects, should be taken into account. However, the association analyses revealed interesting results: The RETSAT and RNMT polymorphisms showed effects on premium cut yields, and the COPA and PALMD polymorphisms showed significant effects on backfat thickness.
Functional candidate polymorphisms
The RETSAT polymorphisms were selected for validation and posterior association analysis due to the biological implication of the RetSat enzyme in retinoid metabolism. RetSat is induced during adipogenesis and is directly regulated by the transcription factor peroxisome proliferator activated receptor alpha and gamma in adipose and hepatic tissues respectively. Moreover, RetSat promotes adipogenesis (Schupp et al. 2009) and lipid accumulation (Moise et al. 2010). Our results agree with the role attributed to RetSat. The RETSAT:g.3320A>C and RETSAT:g.3661delinsT polymorphisms show opposite effects on cut yields and backfat thickness, as expected given that the RetSat enzyme promotes fat deposition. Moreover, the haplotype analysis confirms these effects. The H1 (CinsT) and H2 (CdelT) haplotypes are associated with an increase in premium cut yields and a reduction in backfat thickness. On the other hand, H3 (AinsT) is associated with increases in fat deposition and decreases in premium cut yields. The potential effects of these polymorphisms on mRNA, and protein structure were evaluated using in silico tools. The most interesting corresponded to the RETSAT:g.3320A>C polymorphism, leading to p.Lys173Thr, which is predicted to be tolerated by snap2 and sift tools. However, this polymorphism matched an exon enhancer (regrna prediction tool), whose pattern is GAAGAA (Liu et al. 2003). The SNP would change this pattern to GAAGAC, suppressing the enhancer. RETSAT:g.3661delinsT corresponds to an intronic indel, which could constitute a target site for hsa-miR-1266 (miRNA: 3′-ucGGGACAAGAUGUCGGGACUCc-5′ → target: 5′-acCCTTTTTCCCTTTCTCTGAGa-3′) following the regrna prediction tool. The indel alters the micro-RNA target to 5′-acCCTTTTCCCTTTCTCTGAGa-3′. Although these results are based on predictions, they indicate that these mutations could affect gene expression regulation. However, our gene expression result revealed significant gene expression differences only for the CATA vs. CCTG RETSAT haplotypes and therefore do not fully fit with the obtained phenotypic association results, which may indicate that the analyzed polymorphisms show different levels of linkage disequilibrium with the actual causative mutation.
The RNMT:g.185A>C polymorphism revealed effects on the weights of ham, shoulder and bone-in-loin premium cut yields. The RNMT enzyme is required for cell proliferation processes (Aregger & Cowling 2013) implicated in muscle growth, and consequently, it influences premium cut yields. The evaluated polymorphism leads to p.Gln62Lys, also predicted to be tolerated by snap2 and sift. The regrna prediction tool shows a change in the RNA exposition and accessibility due to the nucleotide substitution (Fig. 4), which could affect post-transcription and protein expression. RNA-Seq gene expression data did not reveal gene expression differences conditional on genotype from liver or hypothalamus, which could support the hypothesis of post-transcriptional effects.

Positional candidate polymorphisms
The COPA:g.48255C>T and PALMD:g.210G>C polymorphisms showed effects on backfat thickness. These genes were selected for further analyses because they both map within QTL regions of SSC4 for backfat thickness (Fernández et al. 2012). Fernández et al. (2012) identified two QTL on SSC4, one at the well-known FAT1 (Marklund et al. 1999; Silva et al. 2011) at 72–91 cM (97–117 Mb) and a second one at 102–109 cM (129–134 Mb) positions. The COPA and PALM genes exactly match those two QTL intervals, at 98 Mb at 129 Mb respectively. The association found for the COPA gene could be a consequence of the linkage disequilibrium with other powerful candidates genes located in this region, such as FABP4 and FABP5, and analyzed in previous studies (Estellé et al. 2006). Validation analysis would be required in order to determine which of these genes (FABP4, FABP5 or COPA) is the actual gene carrying the causal mutation for FAT1.
Despite PALMD:g.210G>C being a synonymous SNP, it could impact gene expression, as shown for the hepatic RNA-Seq gene expression (Table 6). The in silico analysis performed with the regrna tool predicted that this mutation could fall within a transcriptional regulatory motif (Accession no. R0147; Maekawa et al. 1989) in exon 1. The mutation would change the pattern from AGCGGA/TCCGCT to AGGGGA/TCCCCT, affecting gene expression. However, we could not validate the differences in gene expression conditional on the PALMD genotype in a larger sample set. It should be noted that, because the tested PALMD SNP was in a known QTL region (therefore in linkage disequilibrium with the QTL), it is difficult to know the true polymorphism effect and if it actually corresponds to a causal mutation.
Although further functional and genetic validations are needed to prove the association of the polymorphisms identified here, the RETSAT, RNMT and PALMD genes constitute powerful candidates to influence fat deposition and premium cut yields.
RNA editing
Interestingly, RNA editing was detected in the current study when RNA-Seq SNP validation was conducted. The NR3C1:g.4525A>G, COG3:g.13374T>A and ACSM2B:g.13374T>ARNA editing modifications have been validated. Moreover, the same patterns were identified when genotyping was performed in other genetic backgrounds (pure Iberian and Iberian × Large White), indicating that it is a common mechanism.
The most common types of RNA editing modifications in vertebrates are the A-to-I conversion, leading to an A-to-G reading of the cDNA molecule and catalyzed by an adenosine desaminase that acts on RNA family enzymes (ADAR) and the C-to-U conversion, catalyzed by the APOBEC enzyme (Bass 2002; Blanc & Davidson 2003). In the current study the modifications detected were one of the most common conversions: A-to-G (A-to-I in mRNA) or rather A-to-T (A-to-U in mRNA) and T-to-C (U-to-A in mRNA). Functional implications of these modifications were further analyzed by in silico sequence analyses.
The missense modification of COG3:g.4525A>G has been previously described in human, mouse and rat (Shah et al. 2009; Danecek et al. 2012; Holmes et al. 2013) and more recently in chickens (Frésard et al. 2015), proving that at least some RNA editing conservation occurs across vertebrate species. The modification on the component of oligomeric Golgi complex 3 (COG3), a key molecule in protein metabolism, is missense, a p.Ile83Val change, which is functional but tolerated following snap2 and sift prediction tools. The regrna sequence analysis revealed that the modification lies in an exon enhancer region (Accession no. R0815), motif GGAAG, involved in the promotion of alternative splicing (Dirksen et al. 2003), which is likely linked to different biological functions conditional on tissue type, as evidenced in the current results showing different transcripts conditional on tissue type.
The NR3C1:g.102797T>C and ACSM2B:g.13374A>T modifications have not been previously described. The modification in NR3C1, involved in glucocorticoid resistance, lies in the 3′ untranslated region. The regrna prediction tool shows that the NR3C1g.4525 modification could lie in a potential target site for the hsa-miR-2054 miRNA (miRNA: 3′-uuaUUUAAUUUAAAU-------AUAAUGUc-5′ → target: 5′-agaAAGTTGAATTTATAGCTTTTATTGTAc-3′). Finally, the modification on ACSM2B produces p.Ser272Thr. This change is predicted by the sift tool to be tolerated, but regrna predicts a splicing site implicating the ACSM2B:g.13374 position. Moreover, this modification can be considered of major interest because the two different hepatic transcripts showed variations conditional on the tissue that could be functionally related to the H and L animal groups used for the RNA-Seq assay.
The current study has revealed the relevance of the RNA editing, its existence and potential impact, and highlights that RNA editing should be taken into account for future genetic analysis. Furthermore, it could impact the estimates of false discovery rates in SNP validation from RNA-Seq assays due to the differences between the RNA sequence and the DNA region from which it was transcribed.
Here, we have provided valuable SNP data, from over 90 000 SNPs, useful for future studies. Additionally, further approaches can be proposed to validate the promising results obtained, including validations of the polymorphism effects on fatness and premium cut yields and studying the putative functionality of the detected RNA editing modifications.
Acknowledgements
This work was funded by Ministerio de Ciencia e Innovación (MICINN) project AGL2011-29821-C02. Ángel Martínez-Montes was funded by a (FPI) PhD grant from the Spanish Ministerio de Ciencia e Innovación. We wish to thank Fabián Garcia and Anna Castelló for technical assistance, Luis Silió for manuscript corrections and Beatriz Villanueva for comments, suggestions and English style corrections.

