

2025 SELECT ABSTRACTS FROM THE 18th ANNUAL INTERNATIONAL CONFERENCE ON NEONATAL AND CHILDHOOD PULMONARY VASCULAR DISEASE
Corresponding Author:
Jeffrey R. Fineman, MD Professor of Pediatrics
University of California, San Francisco [email protected]
1 Paediatric Chronic Thromboembolic Pulmonary Hypertension (CTEPH): Risk Factors and Outcomes
Anne Vijaykumar, Sadia Quyam, Shahin Moledina
UK Service for Pulmonary Hypertension in Children, Great Ormond Street Hospital for Children, London, UK; Institute of Cardiovascular Sciences, University College London, UK
Chronic Thromboembolic Pulmonary Hypertension (CTEPH) is a rare cause of Pulmonary Hypertension (PH) in children. There is limited data on long-term outcomes following pulmonary endarterectomy (PEA) in this demographic.
We conducted a retrospective analysis of the United Kingdom National Registry for Paediatric Pulmonary Hypertension (2001-2024) identifying 5 children with CTEPH diagnosed through standardised criteria including cardiac catheterisation and cross- sectional imaging.
Among our cohort (60% female, mean age 12 years, range 4-14 years), 80% had identifiable thromboembolic risk factors, predominantly indwelling lines (60%) and thrombophillic conditions (20%). Follow-up duration ranged from 1-5years, with 2 patients followed up for over 4 years. At diagnosis, patients demonstrated reduced exercise capacity (mean six-minute walking distance 361m, range 299-349m) and had echocardiographic evidence of PH (mean TR Vmax 3.67m/s). All patients underwent Cardiac MRI, demonstrating right ventricular (RV) enlargement (mean RV end diastolic volume 109.6ml/m2) and mild-moderate RV systolic impairment (mean RVEF 39.2%). Invasive haemodynamic assessment confirmed significant pressure elevation with mean mPAP 55.6mmHg (range 29-68mmHg) and mean pulmonary vascular resistance index 12.2 WU.m2 (range 5-21WU.m2). Following PEA (100%), improvements were observed in six-minute walking distance (+45m, p=0.0276), RV volumes (-30ml/m2, p=0.0113), RV systolic function (+14.8%, p=0.0031), and haemodynamics (mPAP -28.6mmHg p=0.010; PVRI -11.4WU.m2, p=0.0172). However normalisation of pulmonary haemodynamics to mPAP < 20mmHg was achieved in only 1 patient (20%).
In this paediatric CTEPH cohort, thromboembolic risk factors were highly prevalent. While PEA resulted in significant haemodynamic and functional improvement, complete normalization of pulmonary haemodynamics was uncommon. This suggests residual PH following PEA may be more frequent in paediatric CTEPH compared to adult cohorts. Longitudinal follow-up into adulthood is important to determine long term outcomes in this population.
2 Outcome in a Large Cohort of Infants With Bronchopulmonary Dysplasia Related Pulmonary Hypertension
Caterina Cecilia Lerose, Sadia Quyam, Shahin Moledina
Pulmonary Hypertension Service for Children, Great Ormond Street Hospital NHS Foundation Trust, London, UK; Institute of Cardiovascular Science, University College London, London, UK.
Pulmonary hypertension (PH) is a frequent complication of broncopulmonary dysplasia (BPD) in prematurely born infants. Data are accumulating on the early neonatal implications of BPD-PH however less is known about the natural history thereafter.
We performed a cohort study within a single national referral centre (2001-2025), utilising prospectively acquired data analyse retrospectively. Severity of BPD at 36 weeks gestation and respiratory support at first assessment was graded by Jensen criteria. Severity of PH was graded as per Torok et al.
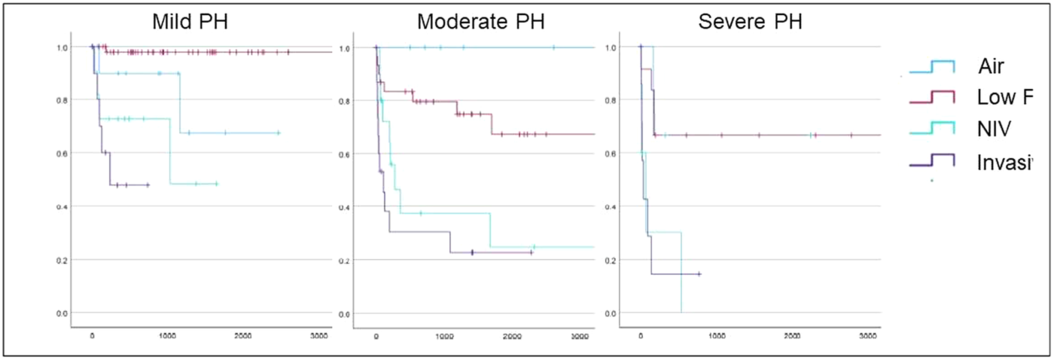
A total of 201 patients with BPD-PH were included at a median age at 1st assessment of 9.1 months [6.4-15.9]. Severity of PH and level of respiratory support at first assessment are summarised in the table. PH severity was weakly correlated with level of respiratory support (Rho 0.29, p< 0.01).
Patients were treated with mono 176 (87.6%), dual 23 (11.4%) or triple therapy 5 (3%).
Over a median 16.1 months follow-up, 78 (38.8%) patients experienced PH resolution, 55 (27.3%) patients died (2 following PH resolution) and 2 (1%) underwent bilateral lung transplantation.
PH severity (HR 2.2, p< 0.01) and level of respiratory support (HR 2.6, p< 0.01) were independently associated with risk of death in a multivariate Cox model. Higher TAPSE values were associated with decreased mortality risk (HR=0.68, p< 0.001). BPD severity showed a trend toward increased mortality risk (HR=1.63, p=0.06), while birth weight and gestational age showed no significant association with mortality.
Infants with BPD-PH experience high rates of PH resolution. Mortality is related to both PH severity and level of respiratory support.
TABLE:
| Air | Low flow O2 | NIV | Invasive | |
|---|---|---|---|---|
| Mild PH | 16 | 57 | 11 | 11 |
| Moderate PH | 7 | 33 | 19 | 16 |
| Severe PH | 3 | 12 | 8 | 8 |
3 High Cardiac Output-Induced Pulmonary Hypertension Due to Kaposiform Hemangioendothelioma in a Neonate
Daniel Beauchamp, Kimberly Miles, Kiersten Ricci, S. Melissa Magness, Michelle Cash, Kimberly Luebbe, Samantha Moore, Russell Hirsch, Paul Critser
Cincinnati Children's Hospital Medical Center, Cincinnati, OH, USA; Department of Pediatrics, University of Cincinnati College of Medicine, Cincinnati, OH, USA
Arteriovenous malformations and vascular tumors can induce PH secondary to high cardiac output. We present a case of neonatal PH due to kaposiform hemangioendothelioma (KHE), a rare non-malignant tumor with high-flow arteriovenous shunting.
A former 32-week male with fetal hydrops was transferred on DOL21 for liver failure. Outside hospital course was also notable for hypoxic respiratory failure requiring intubation, disseminated intravascular-like coagulopathy, gastrointestinal perforations requiring two laparotomies, and PH. On admission, echocardiogram showed normal function, mild-to-moderate mitral regurgitation, and near-systemic RV hypertension which progressed to suprasystemic by DOL25. Liver MRI revealed prominent right iliac vasculature suggestive of pelvic vascular anomaly. MRI and computed tomography confirmed a large high-flow lesion in the posterior pelvis extending into the lower lumbosacral spinal canal, fed predominantly by the right common iliac artery, with IVC dilation and diminutive splanchnic vasculature (suspected “steal” physiology). Given these findings and elevated NTproBNP (> 175,000 pg/mL DOL22), his physiology was most consistent with high output heart failure with PH. On DOL40 he underwent transarterial embolization of multiple branches off the right internal iliac artery and one branch off the medial right external iliac artery. He had suboptimal improvement post- intervention and there was suspicion for vascular tumor, particularly KHE with Kasabach- Merritt phenomenon given his presentation, imaging findings, and complications. Empiric KHE treatment with sirolimus and steroids was started DOL48, and KHE was later confirmed by biopsy. Echocardiographic RV hypertension began to improve around DOL53 and resolved by DOL116. He was weaned off nitric oxide DOL72, extubated DOL83, and weaned to room air DOL101. NTproBNP dramatically improved (30,780 pg/mL DOL55; 9,378 pg/mL DOL63; 1,391 pg/mL DOL114). He remains on sirolimus, and his echocardiogram at 18 months old shows no recurrence of PH.
Vascular anomalies should be considered as causes of neonatal PH. KHE is a rare etiology not previously reported with PH.
4 T-Box Genes Regulate Mesenchymal Cell Fate Specification in the Developing Lung
Kaylie Chiles, Madeline Dawson, Lea Steffes, Casba Galambos, Maya Kumar, Ripla Arora
Michigan State University College of Osteopathic Medicine, East Lansing, MI, USA; Cell & Molecular Biology Program, Michigan State University, East Lansing, MI, USA; Institute for Quantitative Health Science & Engineering, Michigan State University, East Lansing, MI, USA; Department of Obstetrics Gynecology & Reproductive Biology, Michigan State University, East Lansing, MI, USA; Department of Biomedical Engineering, Michigan State University, East Lansing, MI, USA; Division of Pediatric Pulmonary Medicine, Department of Pediatrics, Stanford University School of Medicine, Palo Alto, CA, USA; Department of Pathology, University of Colorado School of Medicine, Aurora, CO, USA.
T-box transcription factor 4 (TBX4) mutations result in neonatal developmental lung disease and pediatric pulmonary arterial hypertension (PAH), indicating an essential role for TBX4 in fetal lung development. TBX4 and its homologue, TBX5, are expressed in fetal lung mesenchyme and are critical for airway branching. However, how loss of these transcription factors affects cell types other than the epithelium embryonically and postnatally has not been evaluated.
We use Tbx4LME-Cre and Tbx4 and Tbx5 conditional alleles to generate lung mesenchyme-specific deletion mouse models. We characterize these mutants using immunofluorescence, in situ hybridization, 3D confocal imaging, and echocardiography.
Complete loss of lung mesenchymal TBX4 causes PAH with right ventricular dilation in adulthood. Concurrent loss of one Tbx5 allele worsens this phenotype. To determine the developmental basis of PAH, we evaluated lungs from Tbx4 and Tbx5 double knockout (DKO) fetuses. As early as embryonic day (E) 12.5, DKO lungs are smaller than controls lungs. At E14.5, we detect ectopic epithelial cells in the sub-mesothelial mesenchyme and at E16.5 and E18.5, we find excess airway smooth muscle and ectopic smooth muscle cells (SMCs) forming pleural bands in the DKO lungs. Due to overlapping location of the ectopic epithelial and SMCs, we postulate that ectopic epithelial cells may give rise to ectopic SMCs. Excess and ectopic SMCs persist postnatally and into adulthood.
We find that loss of T-box gene function gives rise to excess and ectopic SMCs, suggesting that DKO mesenchyme exhibits a propensity for abnormal mesenchymal cell fate specification into smooth muscle subtypes. Using organ and spheroid culture and transcriptomics, our future experiments will assess origin of the DKO ectopic cell populations and the impact of excess SMCs on lung architecture and function.
5 Implantation of the Atrial Flow Regulator (AFR®) Device in Five Patients with Severe Pulmonary Arterial Hypertension: A Promising Bridge to Lung Transplantation
Joseph Pattathu, Robert Dalla-Pozza, André Jakob, Sebastian Michel, Nikolaus Alexander Haas
Division of Pediatric Cardiology and Intensive Care, University Hospital, LMU Munich, Marchioninistraße 15, 81377 Munich, Germany; Division of Pediatric Cardiac Surgery AND Thoracic Surgery University Hospital, LMU Munich, Marchioninistraße 15, 81377 Munich, Germany
Pulmonary arterial hypertension (PAH) is a progressive and life-threatening condition that often leads to right heart failure. For patients in advanced stages, recommended interventions such as atrial septostomy, Potts shunt, or lung transplantation can be critical, though factors like limited donor availability hinder these. In such cases, an effective, safe intervention is needed to bridge the gap until transplantation, improving long-term survival and quality of life. This study explores the potential of the Atrial Flow Regulator (AFR®) device as an interventional bridge in severe PAH.
PAH is characterized by reduced transpulmonary blood flow rather than elevated pulmonary pressures, leading to low cardiac output and symptoms such as syncope. The AFR® device (Occlutech, Germany) creates an intra- atrial shunt, decompressing the right atrium and improving cardiac output while controlling desaturation through the predefined size of the device. In a single-center study, the AFR® was implanted in five patients with severe PAH who were listed for lung transplantation. Device sizes (4 mm, 8 mm, and 10 mm) were selected based on individual anatomical and clinical needs.
Three of the five patients demonstrated significant clinical improvements following AFR® implantation, including improved cardiac output, renal function, and resolution of syncope. These three patients (a 7-year-old girl, a 37- year-old male, and a 39-year-old female) were able to undergo successful bilateral lung transplantation within three months. Despite receiving targeted pharmacological therapy, all patients had worsened clinical status and were awaiting transplantation. These patients experienced improved cardiac function post-implantation, with no further syncopal episodes. The girl, who had been suffering from persistent syncope, has maintained a low NT-proBNP level and remarkable exercise capacity for nearly 8 years post-implantation without transplantation. Additionally, a 21-year-old patient who received an 8 mm AFR® showed no syncope recurrence, with NT-proBNP levels below 200 pg/ml, also without transplantation.
The AFR® device is an effective interventional strategy to stabilize patients with severe PAH and right heart failure, improving cardiac output and providing a bridge to lung transplantation. Proper device sizing is crucial for achieving optimal outcomes. This study highlights the AFR® as a promising option for patients in critical PAH stages, supporting its role in improving long-term outcomes or enabling lung transplantation.
6 Retrospective Review of Safety and Tolerability Endpoints in Young Children Receiving Tadalafil and Ambrisentan for Pulmonary Hypertension
Russell Catania, Ester Liu, Rachel Hopper
Department of Pediatrics, Stanford School of Medicine, Palo Alto; Lucile Packard Children's Hospital Stanford, Palo Alto, CA, USA.
Tadalafil and ambrisentan are once-daily medications used off-label for pediatric pulmonary hypertension (PH) since adult randomized trial data demonstrated superior clinical efficacy in combination compared to monotherapy; however, there is a paucity of data on their use in young children. We hypothesized young children with PH treated with combined tadalafil and ambrisentan tolerated therapy with an acceptable safety profile at our center.
A chart review was performed on 32 patients with World Symposium of PH Group 1, 3, and 5 PH who started tadalafil and ambrisentan between 6 months and 6 years of age. Safety was assessed by mortality, escalation of therapy, incidence of surgery/transplant, hospitalization, and progression to NYHA/Panama functional class (FC) 4. Tolerability was assessed by incidence of adverse drug reactions and treatment discontinuation.
The mean age at initiation was 4.3 years [1.6-6.9 years old]. Median follow-up time was 27.5 months [1.6-118 months]. Two patients (6.2%) died (respiratory failure from pneumonia; out-of-hospital arrest of unclear etiology at 3-year follow up). Four patients (12.5%) started a prostanoid for unsatisfactory hemodynamics. One patient (3.1%) underwent surgical Potts shunt. There were no PH-related hospitalizations, progression to NYHA/Panama FC 4, or lung transplants. One patient (3.1%) permanently discontinued tadalafil due to orthostasis. Common, expected adverse effects included: facial flushing (34%); nasal congestion (25%); headache (19%); nausea/vomiting (15.6%); edema (12.5%); cough, diarrhea (9.4%); epistaxis, orthostasis/lightheadedness (6.3%); anorexia, constipation, penile erection (3.1%). At latest follow-up, 19 patients (59.4%) had no change in FC, 2 patients (6.2%) had improvement in FC, and 1 patient (3.1%) worsened from FC I to II.
Tadalafil and ambrisentan were used in combination safety with an acceptable tolerability profile in young children with PH at our center. A prospective evaluation including efficacy endpoints may further elucidate the utility of these alternative agents.
7 Use of Dupilumab to Treat Cutaneous Complications of Continuous Prostacyclin Infusion
Nidhy Varghese, Erin Ely, Elise Whalen
Department of Pediatrics, Division of Pulmonology, Baylor College of Medicine and Texas Children's Hospital. Houston, Texas, USA; Department of Nursing, Texas Children's Hospital, Houston, Texas USA; Department of Advanced Practice Providers, Division of Pulmonary Medicine, Baylor College of Medicine, Texas Children's Hospital. Houston, Texas, USA
Continuous treprostinil infusion is commonly used for the treatment of advanced or severe pulmonary arterial hypertension (PAH). This infusion can be administered either through a self-implanted subcutaneous catheter or a surgically implanted central venous line. The catheter site is typically secured with adhesive to ensure its stability. However, adhesive-related skin reactions are frequent, and patients with sensitive skin may experience cutaneous exacerbations. Subcutaneous therapy is particularly challenging, as irritation and inflammation at the catheter site can lead to dislodgement, disrupting the therapy and affecting medication absorption.
We report the case of a 10-year-old male with recurrent skin reactions and infections at his subcutaneous treprostinil infusion site. These complications significantly impacted his ability to tolerate the infusion, ultimately affecting the effectiveness of the treatment. Despite trialing different adhesives, infusion was ultimately transitioned to continuous intravenous therapy through a central venous line (CVL) due to concerns for subcutaneous medication absorption. However, the skin reactions reoccurred at the CVL dressing site, requiring frequent changes and increased risk for bloodstream infection. Given the patient's comorbid atopic dermatitis and moderate asthma, dupilumab, a monoclonal antibody that blocks interleukin-4 and interleukin-13 signaling, was introduced to address the chronic skin issues.
Following the first dose of dupilumab, the patient's skin reactions completely resolved. He was able to tolerate the adhesive at his CVL site without any further irritation or complications. The patient continued receiving dupilumab every three weeks, with no recurrence of skin symptoms.
His overall quality of life improved significantly, and he was able to maintain continuous treprostinil therapy without further disruptions.
Dupilumab effectively resolved the chronic adhesive-related skin reactions in this pediatric patient receiving continuous treprostinil infusion. This case underscores the potential benefit of dupilumab in managing skin complications associated with infusion therapies, especially in patients with underlying dermatological conditions such as atopic dermatitis. By resolving these issues, dupilumab allowed the patient to continue treatment without significant disruptions to ensure medication delivery, decrease risk of infection and greatly enhance his quality of life.
8 Neonatal Outcomes in Irreversible Pulmonary Dysplasia Requiring Extracorporeal Membrane Oxygenation: Experience From the Extracorporeal Life Support Organization Registry
Aditya Kalluri Kimberlee Gauvreau K, Jai Khurana, Ravi Thiagarajan, Mary Mullen
Boston Children's Hospital, Boston, MA, USA
Irreversible pulmonary dysplasias (IPDs) are congenital malformations of the pulmonary vasculature such as alveolar capillary dysplasia, often requiring neonatal ECMO and with poor survival. We sought to characterize outcomes for neonates with IPD and PPHN requiring ECMO using the ELSO database. We hypothesized that newborns with IPD would have lower survival, later initiation, and require longer duration of ECMO, and that clinical advances would result in shorter ECMO duration and improved survival in 2011-2021 compared to 2000-2010.
Data were obtained from the ELSO registry from 2000-2021 for neonates aged 0-28 days with ICD-9 and ICD-10 codes defining IPD and PPHN, excluding patients with codes for other pulmonary diseases. Statistical comparisons were made using the Wilcoxon rank sum test for continuous variables or the Fisher's exact test for categorical variables.
There were 3,617 neonates aged 0-28 days requiring ECMO for IPD or PPHN; 84 were included in the IPD group and 3,533 in the PPHN group. Neonates with IPD were more likely to be female, had higher median gestational age by 1 week, and had a higher 5- minute Apgar score (Table 1). Those with IPD were older at the time of ECMO initiation by 1 day (Table 2). Patients with IPD also had a longer duration of ECMO (median 206 hours compared to 134 hours in PPHN). Patients with IPD had a significantly lower rate of survival to discharge than those with PPHN (2.4% vs 70%, P<0.001). Comparison of 2000-2010 and 2011-2021 showed no significant change in age at ECMO initiation or survival in patients with IPD or PPHN.
Neonates with IPD require longer total duration and present with later initiation of ECMO compared to those with PPHN. Despite clinical advances, ECMO duration and survival in IPD have not improved significantly in the last decade.
Table 1: Pre-ECMO characteristics of IPD and PPHN patients
| Total (n=3617) | IPD (n=84) | PPHN (n=3533) | P Value | |
|---|---|---|---|---|
| Sex female (n=84, 3506) | 1451 (40%) | 45 (54%) | 1406 (40%) | 0.018 |
| Birth weight (kg) (n=77, 3300) | 3.2 [2.8, 3.7] (0.2, 7.0) | 3.2 [2.9, 3.5] (1.8, 4.9) | 3.2 [2.8, 3.7] (0.2, 7.0) | 0.29 |
| Gestational age (wk) (n=78, 3297) | 38 [37, 40] (21, 43) | 39 [38, 40] (32, 41) | 38 [37, 40] (21, 43) | 0.013 |
| Apgar score at 5 minutes (n=77, 3193) | 8 [7, 9] (0, 10) | 9 [8, 9] (4, 10) | 8 [7, 9] (0, 10) | < 0.001 |
Values shown are number (percent) or median [interquartile range] (range)
Table 2: ECMO characteristics of IPD and PPHN patients
| Total (n=3617) | IPD (n=84) | PPHN (n=3533) | P Value | |
|---|---|---|---|---|
| Age at ECMO (d) | 2 [1, 3] (0, 28) | 3 [1, 8] (0, 25) | 2 [1, 3] (0, 28) | < 0.001 |
| Mode of ECMO | ||||
| VA | 2521 (70%) | 53 (63%) | 2468 (70%) | 0.090 |
| VV | 954 (26%) | 23 (27%) | 931 (26%) | |
| VV | 4 (< 1%) | 0 (0%) | 4 (< 1%) | |
| A | 116 (3%) | 7 (8%) | 109 (3%) | |
| Conversion | 5 (< 1%) | 0 (0%) | 5 (< 1%) | |
| Other Unknown | 17 (< 1%) | 1 (1%) | 16 (< 1%) | |
| Duration of ECMO (hours) (n=84, 3531) | 135 [91, 211] (1, 2365) | 206 [130, 331] (1, 942) | 134 [90, 209] (1, 2365) | < 0.001 |
| Discharged alive | 2479 (69%) | 2 (2.4%) | 2477 (70%) | < 0.001 |
Values shown are number (percent) or median [interquartile range] (range)
Table 3: Comparison of ECMO characteristics in IPD and PPHN patients by era
| IPD Patients | 2000-2010 (n=40) | 2011-2021 (n=44) | P Value |
|---|---|---|---|
| Age at ECMO (d) | 3 [2, 9] | 3 [1, 7] | 0.47 |
| Duration of ECMO (hours) | 195 [133, 331] | 274 [128, 331] | 0.83 |
| Discharged alive | 0 (0.0%) | 2 (4.6%) | 0.50 |
| PPHN Patients | 2000-2010 (n=2144) | 2011-2021 (n=1389) | P Value |
| Age at ECMO (d) | 2 [1, 3] | 2 [1, 3] | 0.11 |
| Duration of ECMO (hours) | 138 [94, 213] | 126 [85, 198] | 0.002 |
| Discharged alive | 1528 (71%) | 949 (68%) | 0.065 |
Values shown are number (percent) or median [interquartile range] (range)
9 Iloprost Use in Children: Thirteen Years of Experience
Nidhy Varghese, David Spielberg, Neelam Bhatt, Fadel Ruiz, Natalie Villafranco, Raysa Morales-Demori, Elise Whalen, Taylor Saley, Ryan Coleman
Department of Pediatrics, Division of Pulmonology, Baylor College of Medicine, Texas Children's Hospital, Houston, Texas, USA; Department of Pharmacy, Texas Children's Hospital, Houston, Texas, USA; Department of Pediatrics, Division of Critical Care, Baylor College of Medicine, Texas Children's Hospital, Houston, Texas, USA; Department of Advanced Practice Providers, Texas Children's Hospital, Houston, Texas, USA; Department of Pediatrics, Division of Cardiology, Baylor College of Medicine, Texas Children's Hospital, Houston, Texas, USA
Iloprost is a nebulized prostacyclin with rapid onset, short half-life, and few systemic side effects. Reports of use in children with pulmonary hypertension (PH) are limited to cardiac surgical or PPHN case reports. Here we describe a single center experience of inpatient iloprost use for stabilization of the unstable PH patient across the pediatric population.
By retrospective chart review, we identified 329 patients ≤ 18 years of age treated with iloprost from September 2011 – September 2024. Age range was 2 days - 18 years (median 9.5 months) weight range was 0.65 kg – 94 kg (median 8.6 kg), and PH classification spanned all five WSPH groups. Among infants, iloprost was used in infants ranging 22 6/7 – 41 6/7 weeks estimated gestational age (median 35 2/7 weeks). Indication for therapy was often clinical worsening and hemodynamic instability precluding systemic prostacyclin therapy.
Iloprost was nebulized through an inline mesh nebulizer via advanced airway, a non-invasive airway or a handheld ultrasonic nebulizer for the spontaneously breathing patient. Dosing was 0.25-1 mcg/kg/dose for patients < 20 kg and 5 mcg/dose - 20 mcg/dose for patients > 20 kg. Most (n=320) received iloprost on a scheduled basis, typically every 3 hours but as frequently as every 1 hour, while nine received as-needed dosing only. Duration of scheduled therapy was median of 7 days/41 doses (range 1 day/1 dose - 2,792 doses/329 days). Most patients received only one course of scheduled treatment and 12% (41/329) required multiple courses of therapy. No patients required titration of vasopressor support or discontinuation of iloprost therapy. However, Group 2 patients required diuresis for duration of iloprost treatment course. There were no serious adverse events.
Iloprost can be considered for acute stabilization in pediatric PH. Customized administration to meet respiratory needs and interfaces is important to ensure delivery. Iloprost is a safe therapeutic option for the broader pediatric PH population and can be used without adverse events across inpatient settings.
10 Extracorporeal Life Support in Pediatric Pulmonary Hypertension: Support Across the Age and Disease Spectrum
Anna Lang, Corey Chartan, Natalie Villafranco, Taylor Saley, Adam Vogel, Alice King, Enerstina Melicoff, Raysa Morales-Demori, Gazzaneo MC3,6, Jeff Heinle, Yishay Orr, Christopher Rhee, Andrea Ontaneda, Nidhy Varghese, Emmit McKenzie, Ryan Coleman
Baylor College of Medicine School of Medicine, Houston, TX, USA; Division of Pulmonology, Department of Pediatrics, University of Texas at Austin Dell Medical School, Austin, TX, USA; Division of Pulmonology, Department of Pediatrics, Baylor College of Medicine, Houston, TX, USA; Division of Cardiology, Department of Pediatrics, Baylor College of Medicine, Houston, TX, USA; Division of Pediatric Surgery, Department of Surgery, Baylor College of Medicine, Houston, TX, USA; Division of Critical Care Medicine, Department of Pediatrics, Baylor College of Medicine, Houston, TX, USA; Division of Congenital Heart Surgery, Department of Surgery, Baylor College of Medicine, Houston, TX, USA; Pediatric and Congenital Cardiac Surgery, Texas Center for Pediatric and Congenital Heart Disease, University of Texas at Austin Dell Medical School, Austin, TX, USA
Pulmonary hypertension (PH) is a progressive vascular disease characterized by adverse remodeling of the pulmonary vasculature that can occur from a variety of etiologies, all of which adversely affect the right ventricle. Despite medical advances, PH still carries significant risk of morbidity and mortality, and patients may still require aggressive medical support in intensive care units. The use of extracorporeal life support (ECLS) in pediatric PH patients is expanding and its indications are evolving. We describe the utility, diversity of indications, and outcomes of ECLS in children with PH at a single, high-volume center.
Retrospective single-center study of pediatric patients with PH requiring ECLS between 1/2018 and 2/2024. Patients with congenital heart disease-associated PH were excluded. Demographic information, PH medical therapies, ECLS indication, and survival were collected.
Eighty-one patients were identified for analysis: 65 neonates and 16 infants/children. Of the 65 neonates, 60% had a congenital diaphragmatic hernia, 13.8% had persistent pulmonary hypertension of the newborn, 9.2% had meconium aspiration, 6.1% had a congenital pulmonary malformation, 3.1% had lower urinary tract obstruction, and 7.7% had another underlying diagnosis. The median age was ~1 day (IQR 1 - 2 days) and mean weight at cannulation was 3.0kg. The mean time on ECLS was 211.4 hours; 69.2% survived to discharged. Of the 16 infants/children, 50% had idiopathic or heritable pulmonary arterial hypertension, 37.5% had pulmonary veno-occlusive disease, and 12.5% had PH associated with hereditary hemorrhagic telangiectasia. The median age was ~9 years (IQR 1.2 - 14.1 years) and mean weight was 35.3kg. Mean time on ECLS was 457.3 hours; 75.0% survived to discharge. Five were on ECLS as a bridge to medication initiation/optimization; 80% survived to discharge. Seven patients were on ECLS as a bridge to lung transplantation; 86% survived to discharge.
ECLS can safely support pediatric PH patients of all ages with various disease etiologies, can facilitate the initiation of PH-targeted pharmacotherapy, and can be used as a bridge to transplant or recovery.
11 Lung Ultrasound Scores in Extremely Preterm Infants With Evolving Bronchopulmonary Dysplasia: Effect of Dexamethasone Treatment
Joshua Hazan Mea, Phoenix Plessas-Azurduy, Thomas Sonea, Pasinee Kanaprach, Carolina Michel Macias, Shiran Moore, Punnanee Wutthigate, Jessica Simoneau, Daniela Villegas Martinez, Andreanne Villeneuve, Anie Lapointe, Guilherme Sant'Anna, Gabriel Altit
Division of Neonatology, Faculty of Medicine and Health Sciences, McGill University, Montreal, QC, Canada; Faculty of Medicine, Université de Montréal, Montreal, QC, Canada; Division of Neonatology, Department of Pediatrics, Montreal Children's Hospital, Montreal, QC, Canada; Faculty of Medicine, Universidad Autónoma de Querétaro, Querétaro, Mexico; Division of Neonatology, Department of Pediatrics, Dana Dwek Children's Hospital, Tel Aviv Sourasky Medical Center, Tel Aviv, Israel; Department of Pediatrics, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand; Division of Neonatology, Department of Pediatrics, CHU Sainte-Justine, Université de Montréal, QC, Canada
Bronchopulmonary dysplasia (BPD) is a common complication in premature infants, often linked to prolonged mechanical ventilation. Dexamethasone (DEXA) is used in these neonates to reduce pulmonary inflammation and facilitate the transition to non-invasive respiratory support. Lung ultrasound (LUS) can assess lung parenchymal changes, particularly variations in density related to atelectasis and inflammation. We hypothesize that LUS scores will decline following DEXA administration in preterm infants with evolving BPD.
This prospective observational study enrolled preterm infants born at < 29 weeks gestational age (GA) who received DEXA for evolving lung disease. LUS was performed at DEXA initiation, days 3, 7, and 14 of treatment, 1 and 2 weeks post-treatment, and at 36 weeks corrected GA. Each of the six lung regions was scored by a blinded reviewer from 0 to 3, with a maximum possible score of 18 indicating greater consolidation.
From 2021 to 2024, 38 neonates were recruited (61% male, 79% inborn), with 81% developing moderate-to-severe BPD as per the Canadian Neonatal Network.
Three patients died (8%). The mean GA at birth was 25.4 weeks (SD: 1.4), and the mean birthweight was 757g (SD: 192g). Median Apgar scores were 6 (IQR: 5-7) and 8 (IQR: 6-8) at 5 and 10 minutes, respectively. Among survivors, the average hospitalization duration was 134 days (SD: 47). A mixed-effects model demonstrated a significant decline in average LUS score (β = -0.54, 95% CI: -0.67 to -0.40, p < 0.001) and in percentage scores (β = -3.0, 95% CI: -3.7 to -2.2, p < 0.00001) during DEXA treatment. The decline began with DEXA exposure and continued without rebound after cessation, with the lowest values observed at 36 weeks corrected GA.
DEXA treatment was associated with a sustained reduction in LUS scores, suggesting improved aeration, with benefits lasting beyond treatment cessation.
12 Pulmonary Hypertension Outcomes After Closure of Atrial Septal Defect in Infants With Bronchopulmonary Dysplasia or Congenital Diaphragmatic Hernia: A Retrospective Review
John Wiegand, James Thompson, Bruce Landeck, Jamie Fierstein, Dina Ashour, Joana Machy, Amy Kiskaddon, Marisol Betensky, Grace Freire
Department of Pediatrics, University of South Florida Morsani College of Medicine, Tampa, FL, USA; Department of Pediatrics, Divisions of Pediatric Cardiology, Clinical and Translational Research, Neonatology, Hematology-Oncology Johns Hopkins All Children's Hospital, St. Petersburg, FL, USA
Bronchopulmonary Dysplasia (BPD) and Congenital Diaphragmatic Hernia (CDH) are associated with pulmonary hypertension (PHTN), which can worsen with atrial septal defect (ASD) due to increased pulmonary blood flow. Early transcatheter ASD closure (≤ 1 year of age) may reduce PHTN severity, but its effects in infants with BPD and/or CDH are unclear. This study investigated the impact of early ASD closure on PHTN severity and whether ASD size, gestational age, and baseline PHTN severity influenced post-procedural medication duration.
A retrospective, single-center cohort study included 16 infants with BPD and/or CDH and PHTN who underwent early transcatheter ASD closure (January 2021–September 2024). Outcomes—PHTN severity, respiratory support, and medication use—were assessed at baseline, 24 hours, and 1-, 3-, 6-, and 12-months post-procedure. Paired Z- tests and Wilcoxon Signed-Rank tests analyzed differences over time. Associations between medication duration and clinical factors were assessed using odds ratios and 95% confidence intervals (p < 0.05).
Within 12 months, 3 (25%) infants with moderate PHTN and 3 (75%) infants with mild PHTN improved to normal, with 10 (63%) infants showing overall improvement.
Vasodilator and diuretic dosages decreased by 48% and 92%, respectively. Invasive respiratory support decreased by 57%, non-invasive support by 83%, and 6 (40%) infants transitioned to room air. A larger ASD (> 5 mm vs. ≤ 5 mm) was associated with three times longer diuretic use, and an earlier gestational age (≤ 32 weeks vs. > 32 weeks) was linked to twice the duration of diuretic use. Higher baseline PHTN severity prolonged diuretic use by 1.2 times, but none of these factors affected vasodilator duration.
PHTN severity, respiratory support, and medication use improved following early transcatheter ASD closure in infants with BPD and/or CDH. Larger ASD, earlier gestational age, and higher baseline PHTN severity were associated with prolonged diuretic use. Further research is needed to refine optimal timing for ASD closure.
13 Serial Pulmonary Artery Pulsatility and Compliance as Simple Reproducible Echocardiographic Parameters to Guide Management in Pediatric Pulmonary Arterial Hypertension
Katelyn Benson, Usha Krishnan
Pediatric Cardiology, Columbia University Irving Medical Center, Morgan Stanley Children's Hospital of New York Presbyterian, New York, New York, USA
Serial pulmonary artery (PA) measurement by echocardiogram is an easily reproducible noninvasive parameter that can be trended over time to guide management of pulmonary arterial hypertension (PAH).
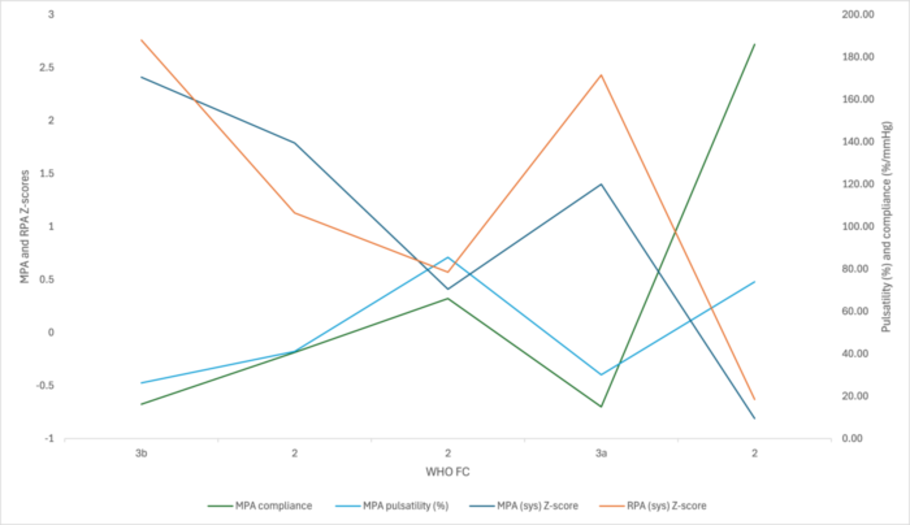
Data from 25 patients with PAH ages (3 months–18 years) seen between 2013-2025 were collected by retrospective chart review. Main and right PA (MPA, RPA) dimensions, pulsatility, compliance, and indirect measures of PAH severity were measured on echocardiograms at 4 time points (corresponding with hemodynamic studies) at baseline, 6-12 months of therapy, escalation of disease and most recent visit. Largest and smallest MPA and RPA diameters in a cycle were charted and converted to Z-scores using Boston Z-score calculator. PA compliance and pulsatility were calculated. These will be correlated with concurrently documented clinical status, hemodynamic data, exercise parameters and biomarkers (proBNP).
The table below describes pilot data on 5 patients. A complete analysis of 25 patients will be presented. A graphic on 1 patient depicts positive correlation between clinical status, pulsatility and compliance, and negative correlation with PA size.
These preliminary data suggest that PA size, pulsatility and compliance correlate with hemodynamic, biomarker, and functional data in children with PAH. Echocardiographic evaluation of PA pulsatile function potentially represents a simple, reproducible and non-invasive metric to assist in managing these complex patients and provide additional information on response to therapies.
| TIMEPOINT1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| WHOFC | MPA (sys) Z-score | RPA (sys) Z-score | MPAcompliance (%/mmHg) | MPA pulsatility (%) | PVR(mmHg) | PVR/SVR ratio | proBNP(pg/ml) | 6MWD(m) | |
| Patient 1 | lllb | 2.41 | 2.76 | 16.19 | 26.27 | 28.80 | O.BO | 293 | 367 |
| Patient2 | IV | 5.17 | 3.31 | 47.23 | 92.45 | 19.50 | 0.79 | 3426 | 300 |
| Patient3 | IV | 5.63 | 2.85 | 2.92 | 7.76 | 30.00 | 1.59 | 501 | 420 |
| Patient4 | II | 5.51 | 2.10 | 16,65 | 37.36 | 28.00 | 1.09 | 1831 | 450 |
| Patient5 | II | 5.22 | 5.85 | 15.24 | 31.82 | 12.11 | 0,94 | 334 | 121 |
| MEAN | 4.79 | 3.37 | 19,65 | 39.13 | 23.68 | 1.04 | 1277 | 332 | |
| SD | 1.34 | 1.45 | 16.44 | 31.82 | 7,68 | 0,33 | 1359 | 131 | |
| TIMEPOINT2 | |||||||||
| Patient 1 | II | 1.79 | 1.13 | 40.66 | 41.10 | 6.20 | 0.68 | 27 | 458 |
| Patient2 | II | 5.48 | -0.43 | 80,85 | 130.25 | 7.00 | 1.10 | 156 | 327 |
| Patient3 | 1-11 | 1.78 | 1.65 | 8.28 | 19.73 | 13.00 | 0,96 | 81 | 565 |
| Patient4 | I | 4.28 | 3.39 | 60,84 | 67.00 | 8.30 | 0.70 | 31 | 467 |
| Patients | II | 4.33 | 3.01 | 5.90 | 7.94 | 7.75 | 0.52 | 69 | 298 |
| MEAN | 3.33 | 1.44 | 47.66 | 64.52 | 8.63 | 0.86 | 74 | 454 | |
| SD | 1.67 | 1.53 | 32.67 | 48.59 | 2.66 | 0.23 | 52 | 110 | |
| TIMEPOINT 3 | |||||||||
| Patient 1 | II | 0.41 | 0.57 | 66,07 | 85.52 | 12.90 | 0.57 | 36 | 509 |
| Patient2 | II | 4.68 | 4.27 | 35,17 | 58.82 | 11.00 | 1.01 | 163 | 402 |
| Patient3 | 1-2 | 2.98 | 1.02 | 6.11 | 16.46 | 34.20 | 1.36 | 357 | 596 |
| Patient4 | I | 3.32 | 1.14 | 17.90 | 17.13 | 8.38 | 0.60 | 41 | 233 |
| Patient5 | II | 3.20 | 5.53 | 13,69 | 20.69 | 13.82 | 0.67 | 84 | 393 |
| MEAN | 2.92 | 2.51 | 27.79 | 39.72 | 16.06 | 0,84 | 136 | 427 | |
| SD | 1.55 | 2.24 | 23,91 | 31.13 | 10.35 | 0,34 | 134 | 137 | |
| TIMEPOINT 4 | |||||||||
| Patient 1 | Illa | 1.40 | 2.43 | 14.99 | 30.10 | 22.37 | 1.12 | 168 | 454 |
| Patient2 | II | 3.83 | 1.70 | 12.82 | 33.14 | 25.00 | 1.09 | 2211 | 392 |
| Patient3 | II | 1.90 | 0.32 | 32.90 | 111.80 | 15.61 | 1.15 | 60 | 592 |
| Patient4 | II | 2.28 | 4.06 | 96.90 | 73.42 | 6.41 | 0.41 | 57 | 452 |
| Patient5 | II | 2.15 | 0.91 | 0.00 | 47.52 | 9.50 | 0.78 | 36 | 472 |
| MEAN | 2.31 | 1.88 | 31.52 | 59.20 | 15.78 | 0.91 | 506 | 472 | |
| SD | 0.91 | 1.45 | 38.38 | 34.03 | 8.00 | 0.32 | 954 | 73 | |
14 Expanding the Phenotypic Spectrum of MECOM-Associated Syndrome: Rare Variants Cause Early-Onset Syndromic Pulmonary Arterial Hypertension
Carrie Welch, Meriel McEntagart, Shahin Moledina, Carrie Morgan, Emilia Swietlik, Chau Hou, Lu Qiao, Emily Callejo, Savanna Craib, Emilia Bijlsma, Patrice Bouvagnet, Tamir Dagan, Jacqueline Eason, Francis Flinter, Aakash Joshi, Jeremie Mortreux, Fadel Ruiz, Deborah Shears, Celia Azevedo Soares, Nidhy Varghese, Wendy Chung
Boston Children's Hospital, Boston, MA, USA; Harvard Medical School, Boston, MA, USA; St George's University Hospitals NHS Foundation Trust, London, UK; Cambridge University Hospitals NHS Foundation Trust, Cambridge, UK; Columbia University Irving Medical Center, New York, NY, USA; Great Ormond Street Hospital, London, UK; Leiden University Medical Center, Leiden, NE; Eurofins Biomnis, France; University Hospital of Martinique, Fort de France, France; Schneider Children's Medical Center, Petah Tikva, Israel; Nottingham University Hospitals NHS Trust, Nottingham, UK; Guy's & St Thomas’ NHS Foundation Trust, London, UK; Oxford University Hospital NHS Foundation Trust, Oxford, UK; Texas Children's Hospital, Houston, TX; Baylor College of Medicine, Houston, TX Unidade Local de Saúde Santo António, Porto, Portugal
Heterozygous de novo variants, often in developmental patterning genes, are the most frequent genetic cause of pediatric pulmonary vascular diseases. De novo variants in MECOM, a developmental and hematopoietic transcription factor, cause a rare early-onset and heterogeneous syndrome associated with bone marrow failure, skeletal abnormalities, and other congenital anomalies. We previously identified MECOM as a candidate pediatric pulmonary arterial hypertension (PAH) gene using trio exome sequencing and hypothesized that PAH is a pulmonary manifestation of MECOM-associated syndrome.
To test the role of MECOM in pediatric PAH and further define the clinical phenotype of MECOM-associated syndrome, we queried GeneMatcher and screened rare disease databases for individuals with predicted deleterious MECOM variants. We performed clinical characterization of patients, protein modeling of genetic variants, and pulmonary expression analyses.
We identified fourteen individuals with rare deleterious MECOM variants, including 11 unrelated probands (8 de novo, 2 inherited, 1 unknown). The variants included both protein-truncating (n = 4) and missense variants (n = 7). Ten individuals had severe or mild thrombocytopenia, seven had skeletal issues, at least six had PAH associated with cardiac anomalies, one had idiopathic PAH, and nine had other medical conditions. Three were diagnosed in utero and died in the neonatal period. All missense variants mapped to the zinc finger 6 or zinc finger 8/9 region, a hotspot for MECOM- associated syndrome. 3D protein modeling predicted that both regions are DNA- binding, and that the variants may interfere with binding to a VEGFR2/KDR enhancer. Data from LungMAP showed that MECOM is primarily expressed in pulmonary arterial endothelial cells.
These data support a causal role for MECOM variants in early-onset syndromic PAH and indicate that PAH monitoring should be considered for all individuals with rare MECOM variants. The pathogenetic mechanism for PAH and cardiac defects may be impaired VEGFR2/KDR signaling.
15 Metabolic Carts in Pediatric Pulmonary Hypertension
Sierra Casey, Cathy Sheppard, Susan Richards, Maryann McMaster, Stephanie Lavoie, Angela Bates
Department of Pediatrics, Division of Pediatric Cardiology, University of Alberta, Edmonton, Alberta, Canada
Pediatric pulmonary hypertension (PH) patients have decreased growth parameters compared to healthy children. Chronic disease research shows increased resting energy expenditure (REE) negatively impacts growth. Increased weight in pediatric patients with PH correlates with improved Panama functional classification, which suggests that there is a relationship between PH severity and REE. Metabolic carting measures gas exchange to calculate REE. We hypothesized that REE in pediatric PH patients, measured with metabolic carts, would be higher than predicted for weight and height, and that a higher relative REE would correlate with increased disease severity.
14 outpatient pediatric PH patients had REE calculated using metabolic carts. Calculated REE was compared to predicted REE for weight and height. Patients’ Functional Classification and 6-minute walk distance (6MW) were collected from chart review and correlated with measured and predicted REE.
Patients were 50% male and average age was 11.6 years (range: 3.8-16.8). 12/14 were NICE classification group I, 1/14 was group II, and 1/14 was group 3. Mean weight Z- score was -0.48, height Z-score was 0.11, and BMI Z-score was -0.8. 4/14 patients were on home oxygen. Panama functional classification was I/II in 8/14 and IIIa/IIIb in 6/14. 10/14 patients completed 6MW with average of 72% predicted distance for age.
Measured REE on average was 99% predicted (range: 75-121%). Patients with Functional Classification group I/II had on average 95% predicted REE while patients with functional classification IIIa/IIIb had on average 104% predicted REE, although results were not statistically significant (p=0.25).
Metabolic carts are a safe and feasible way to estimate energy requirements in pediatric PH patients. Challenges in recruitment occurred related to poor compliance in young patients and exclusion of those requiring oxygen supplementation during testing. A larger multi-center study could demonstrate further utility of metabolic carts in pediatric PH.
16 Association of Portosystemic Shunting With Pulmonary Arterial Hypertension in Patients With Heterotaxy Syndrome
Thomas Conrads, David Schidlow, Mary Mullen
Department of Pediatrics, Division of Cardiology, Lucile Packard Children's Hospital, Stanford University School of Medicine, Palo Alto, CA, USA; Department of Cardiology, Boston Children's Hospital, Harvard Medical School, Boston, MA, USA
Heterotaxy syndrome is a rare disorder characterized by the abnormal location of the abdominal and/or thoracic organs. One uncommon diagnosis that has been previously described in heterotaxy is pulmonary hypertension (PAH). In this study, we describe a large pediatric cohort with heterotaxy and PAH and specifically compare the severity of PAH in the population with heterotaxy and portosystemic shunt.
We conducted a search in Boston Children's Hospital database for patients diagnosed with heterotaxy and PAH who underwent cardiac catheterization from 2000-2024. Variables analyzed were gender, cardiac position, isomerism, status of the IVC, spleen status and presence of portosystemic shunt. Each independent variable was analyzed with pulmonary vascular resistance (PVR) and mean pulmonary artery pressure (mPAP).
A total of 53 patients with variety of ages, anatomy and surgical repair status met the inclusion criteria. The mean mPAP was 32.69mmHg, and the mean PVR was 4.83iWUxm2. Multivariable regression analysis demonstrated that portosystemic shunt was significantly associated with increased PVR (p=0.010) and elevated mPAP (p=0.011). Polysplenia was also associated with higher mPAP (p=0.034). Cardiac position, sex, isomerism type, and IVC status, were not significantly associated.
Subgroup analysis showed that patients with both portosystemic shunt and polysplenia had significantly higher mPAP (53.4mmHg) and PVR (16.04iWUxm2) compared to the rest (mPAP: 30.57mmHg, PVR: 3.69iWUxm2).
Our study represents an analysis of PAH in a large cohort of patients with heterotaxy undergoing cardiac catheterization. We identified a significant association between portosystemic shunting and increased PVR and mPAP. These findings suggest that abnormal hepatic venous return may contribute to the pathogenesis of PAH in heterotaxy. These results support the need for early screening and intervention strategies in patients with heterotaxy and portosystemic shunting. Further research should explore whether interventional management of portosystemic connections could reduce pulmonary hypertension progression and impact therapeutic response.
17 Pulmonary Hypertension After Hematopoietic Cell Transplantation Presenting as Cardiorespiratory Failure in Infants With Hemophagocytic Lymphohistiocytosis
Anthony Williams, Ahmad Rayes, Ryan Carpenter, Liezi Domingo
University of Utah Primary Children's Hospital Salt Lake City, UT, USA
Pulmonary hypertension (PH) is a rare but potentially fatal morbidity following hematopoietic cell transplant (HCT) for hemophagocytic lymphohistiocytosis (HLH). However, there is limited literature about the incidence and management of PH after HCT.
We report the case of two infants presenting with acute cardiorespiratory failure and severe PH after HCT for primary HLH which was complicated by hepato-renal venoocclusive disease (VOD) and thrombotic microangiopathy (TMA) who were successfully treated with combination PH therapy.
Case 1 is an 11-month-old female who had acute cardiorespiratory decompensation ~3.5 months after HCT. Echocardiogram revealed severe PH with right ventricular dysfunction. Inhaled nitric oxide (iNO), bosentan and intravenous treprostinil were initiated. Cardiac catheterization showed a mPAP 47 mmHg and PVRi 11.5 WU*m2. She was treated with triple targeted PAH therapies with subcutaneous treprostinil, sildenafil and bosentan, as well as eculizimab for TMA.
Case 2 is a 7-month-old male who developed respiratory failure ~ 2 months after HCT and found to have severe suprasystemic PH on echocardiogram. He was started on iNO, bosentan, and iloprost, and defibrotide for TMA with good response. Cardiac catheterization performed after stabilization revealed a mPAP 24 mmHg and PVRi 3.46 WU*m2. He was switched to sildenafil, bosentan and selexipag on discharge.
‘Due to hemodynamic instability, both patients were started on up-front combination PH ‘therapies with clinical improvement. They tolerated weaning off all PH therapies about a year ‘after diagnosis (12-15 months). Serial echocardiograms and repeat hemodynamics showed normal ‘mPAP and PVRi. No adverse effects from PH therapies were observed in either case.
‘PH after HCT can lead to rapid cardiorespiratory decompensation. Prompt diagnosis and ‘aggressive treatment with targeted PH therapy allow normalization of hemodynamics and ‘improve outcomes.
18 Pulmonary Vascular Disease, Pulmonary Arteriovenous Malformations and High Cardiac Output: Searching for a Unifying Diagnosis
Lillam Aquino, Elizabeth Colglazier, Claire Parker, Elena Amin, Jeffrey Fineman, Hythem Nawaytou
University of California, San Francisco, Benioff Children's Hospital, San Francisco, CA, USA
This case involves a child lacking a unifying diagnosis for her presentation that includes pulmonary vascular disease (PVD).
TA recently emigrated from Yemen at 8.5 years of age. She presented with short stature, developmental delay, central hypothyroidism, patent ductus arteriosus (PDA), and PVD on sildenafil. She had intermittent exertional central cyanosis and chest pain. Her pre- and post-ductal oxygen saturations were 100%/96% at rest and 89%/91% after a six- minute walk test.
Initial echocardiogram revealed a moderate PDA with left-to-right shunting, holodiastolic flow reversal in the abdominal aorta, normal biventricular function, and a PFO with left- to-right shunting. Hemodynamic evaluation showed high but equal pulmonary (Qp) and systemic (Qs) blood flow (7.3L/min/m²), pulmonary venous desaturation (86%), and elevated pulmonary artery pressures with high left atrial pressure (14mmHg), leading to an indexed pulmonary vascular resistance (PVRi) of 4.5WU. Acute vasodilator testing showed significant vasoreactivity (Qp: 11.6L/min, Qp:Qs 3.5, PVRi 2.5WU).
Four days after initial cardiac catheterization, she experienced a left MCA stroke, and brain MRI suggested congenital cerebral vasculopathy. Genetic testing was notable for variant of unknown significance in the mitochondrial genome, MT-ND4 (NC_012920.1) m.10868A>G (p.I37V). Further workup also revealed central adrenal insufficiency and Von Willebrand disease. Her PDA was closed one month later, and she remained on Tadalafil but continued to experience exertional cyanosis. A follow-up hemodynamic assessment a year later showed a high cardiac index (7.1L/min/m²), low PVRi (2.2WU), and pulmonary venous desaturation, along with pulmonary micro arteriovenous malformations. No evidence of liver disease or hereditary hemorrhagic telangiectasia was present. Despite discontinuing Tadalafil, a repeat evaluation 1.5 years later showed similar findings. Cardiac MRI confirmed a high-output state with biventricular dilation.
This case represents a child with mixed presentation of pulmonary AVM, elevated PVR, high cardiac output without a unifying diagnosis. We are seeking expert opinions on further work-up and treatment for this child.
19 Management of Pulmonary Vein Stenosis Following an Institutional Protocol of Aggressive Surveillance and Intervention and Medical Therapy With Sirolimus
Anna Lampe, Lauren Sanlorenzo, Dominder Kaur, Christopher Petit, Usha Krishnan
Divisions of Cardiology, Neonatology, Hematology/Oncology/Stem Cell Transplant, Department of Pediatrics, Columbia University Vagelos College of Physicians and Surgeons and NewYork-Presbyterian Morgan Stanley Children's Hospital, New York, NY, USA
Pulmonary vein stenosis (PVS) has a high morbidity, mortality and recurrence rate after anatomic intervention, although outcomes have been improving recently. Sirolimus has shown promise in treating PVS, but there is a relatively small body of literature on its use. This study describes the background, treatment, and outcomes of the pediatric PVS cohort at our institution treated with sirolimus.
All patients had multivessel PVS, underwent a protocol of close surveillance with echocardiograms, CT angiograms and aggressive intervention with angioplasty, stenting and/or surgery and received sirolimus (n=26). All patients were treated with sirolimus between 2021 and 2025. The current study is a retrospective cohort review and there is a follow up plan of comparison with a historic cohort of PVS patients from 2010-2020.
The median gestational age was 30 weeks (27-37 weeks) and median birth weight was 1050 g (624-2720g). Most (62%) have bronchopulmonary dysplasia and 44% have complex congenital heart disease (CHD). All had multivessel disease with a median of 3 stenotic veins (2.8-4). Patients underwent multiple procedures yearly (median 2.5, 1.4- 4.2). This cohort's hospitalization burden is high, with median hospitalizations per year at 2.7 (2.1-4.1), and 1.7 requiring ICU stay (0.7-2.4 days). Three patients died(11.5%), 2 from severe viral illness and 1 from complex CHD with heart failure. The target serum sirolimus level was 8-12 ng/mL. Sirolimus was paused in 46% for viral illness or procedures for a range of 4-14 days. In 8%, sirolimus was stopped for side effects (1 poor appetite and irritability and 1 frequent respiratory infections). 12% (n=3) stopped sirolimus at 4-5 years old after resolution of PVS.
In the modern era of aggressive surveillance, intervention and therapy with sirolimus, survival for patients with severe multiple vein stenosis has improved. Sirolimus was well tolerated with an acceptable side effect profile.
| 25th | |||||
|---|---|---|---|---|---|
| Table 1: | Median percentile 75th percentile n | Percent | |||
| Total | 26 | 100 | |||
| Gestational Age, weeks | 30 | 27 | 37 | ||
| Female sex | 8 | 30.8 | |||
| Birth weight, g | 1050 | 624 | 2720 | ||
| Race/ethnicity | |||||
| White, Non-Hispanic | 4 | 15.4 | |||
| Black, Non-Hispanic | 4 | 15.4 | |||
| Hispanic (any race) | 9 | 34.6 | |||
| Asian, Non-Hispanic | 2 | 7.7 | |||
| Other | 4 | 15.4 | |||
| Prefer not to disclose | 3 | 11.5 | |||
| Non-PVS complex | |||||
| congenital heart disease | 11 | 44 | |||
| Lung disease | |||||
| Bronchopulmonary | |||||
| dysplasia (BPD) | 16 | 61.5 | |||
| Grade 1 BPD | 0 | 0 | |||
| Grade 2 BPD | 9 | 34.6 | |||
| Grade 3 BPD | 5 | 19.2 | |||
| BPD, unable to determine grade (outside hospitalrecords) | 2 | 7.7 | |||
| 25th | |||||
|---|---|---|---|---|---|
| Table 2: Outcomes | Median percentile 75th percentile n | Percent | |||
| Total | 26 | 100 | |||
| Length of follow up, weeks | 137.4 | 47.4 | 215.2 | ||
| Arrest | 12 | 48 | |||
| ECMO | 4 | 15.4 | |||
| Mortality | 3 | 11.5 | |||
| Hospitalizations: | 7.5 | 4 | 12 | ||
| Total hospitalizations since PVS diagnosis | |||||
| Average hospitalizations | |||||
| per year since PVS | |||||
| diagnosis | 2.7 | 2.1 | 4.1 | ||
| Total hospitalizations since | |||||
| PVS diagnosis requiring | |||||
| ICU | 3.5 | 1.25 | 4.8 | ||
| Average hospitalizations | |||||
| per year since PVS | |||||
| diagnosis requiring ICU | 1.7 | 0.7 | 2.4 | ||
20 Sotatercept Use in Pediatric Patients With Potts Shunt
Kelly Merrill, Angela Drussa, Emma Jackson, Anne Davis, Meredith Riker, Manish Aggarwal, Mark Grady, Delphine Yung
Seattle Children's Hospital, University of Washington, Seattle, WA, USA; Washington University School of Medicine, St. Louis, MO, USA
Sotatercept is an activin signaling inhibitor approved by the FDA in March 2024 to treat adults with pulmonary arterial hypertension (PAH). The adult STELLAR trial showed that adding Sotatercept to baseline therapy led to improvement in PAH; however, serious bleeding events and the risk for severe thrombocytopenia were reported1. The palliative Potts shunt, a connection between the left pulmonary artery and descending aorta, has been performed in children with suprasystemic PAH since 20032 to protect right ventricular function at the expense of lower body oxygen desaturation. Patients with a Potts shunt, or any hemodynamically significant open shunts, were excluded in all sotatercept trials due to concerns for bleeding. However, we hypothesized that sotatercept would be an effective and safe adjunct therapy in adolescents on triple PAH therapy and status post Potts shunt.
Four adolescent patients with PAH and Potts shunt received sotatercept in addition to triple therapy (Table 1). Prior to starting the medication, patients and families were counseled on the possible side effects of sotatercept, including the increased risk of bleeding. The medication package insert was followed for dosage and lab monitoring.
All patients completed at least 6 doses of sotatercept and uptitrated to 0.7 mg/kg. Hemoglobin and platelets showed little change and remained in normal range for age.
One patient, with a long history of mild epistaxis, experienced a slight increase in epistaxis frequency after initiation of treatment. All patients had mild decrease in B-type natriuretic peptide. 3 had higher 6MWT, 3 had improved TAPSE, 1 had improvement in functional class and 2 had decrease in estimated RV pressure by TRJ. (Table 2 & 3).
We conclude that the addition of sotatercept in adolescent patients with PAH and Potts shunt appears safe during early dosing. There are early signs of improvements in functional class and estimated RV pressure.
Table 1: Demographics
| CASE 1 | CASE 2 | CASE 3 | CASE 4 | |
|---|---|---|---|---|
| AGE (YR) | 13 | 12 | 13 | 16 |
| WEIGHT (KG) | 39.5 | 38.7 | 75 | 31 |
| WHO classification | GROUP 1 | GROUP 1 | GROUP 1 | GROUP 1 GROUP 3 |
| CONCURRENT PAH THERAPIES | Tadalafil 30 mg QD Ambrisentan 10 mg QD Selexipag 1,600 mcg BID |
Sildenafil 20 mg TID Ambrisentan 10 mg QD Treprostinil 120 ng/kg/min |
Tadalafil 40mg QD Macitentan 10 mg QD Treprostinil 111 ng/kg/min |
Tadalafil 20 mg QD Ambrisentan 5 mg QD Treprostinil 76 ng/kg/min |
POTTS SHUNT SURGERY YEAR |
2022 | 2021 | 2016 | 2018 |
| COMPLICATIONS | Patient had history of acquired Von Willebrand with mild epistaxis, occurring on average every 2 months. Frequency of epistaxis increased post initiation; however, the overall frequency was still low (< 1x per month). |
None | Pain/anxiety with SQ injections After dose #3, complained of fatigue, SOB, nasal congestion, and anorexia. Labs, 6MWT and echo stable. No evidence of telangiectasias per ENT but nasal inflammation treated with nasal steroids. Macitentan, started 2 months prior, may have contributed to nasal congestion. |
None |
Table 2: Labs prior to receiving dose
| SOTATERCEPT AT Q3 WK INTERVALS | CASE 1 | CASE 2 | CASE 3 | CASE 4 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dose mg/kg | HGB | PLT | NT-P BNP | Dose mg/kg | HGB | PLT | NT-P BNP | Dose mg/kg | HBG | PLT | BNP | Dose mg/kg | HBG | PLT | BNP | |
| DOSE 1 | 0.3 | 14.8 | 196 | 484 | 0.3 | 15.4 | 242 | 475 | 0.3 | 14.9 | 269 | 48 | 0.3 | 11.9 | 176 | 152 |
| DOSE 2 | 0.3 | 16.3 | 174 | -- | 0.3 | 16 | 243 | 276 | 0.7 | 14.4 | 247 | 15 | 0.7 | 13.4 | 144 | 148 |
| DOSE 3 | 0.7 | 16.1 | 196 | 514 | 0.7 | 16.9 | 200 | 301 | 0.7 | 15.2 | 221 | < 5 | 0.7 | 13.6 | 171 | 109 |
| DOSE 4 | 0.7 | 14.6 | 188 | 312 | 0.7 | 15.8 | 216 | 266 | 0.7 | 14.2 | 224 | 22 | 0.7 | 15.3 | 180 | 57 |
| DOSE 5 | 0.7 | 14.8 | 182 | 360 | 0.7 | 15.8 | 219 | -- | 0.7 | 14.6 | 230 | 7 | 0.7 | 14.5 | 129 | 129 |
| DOSE 6 | 0.7 | 15.3 | 210 | 403 | 0.7 | 15 | 230 | 81 | 0.7 | -- | -- | -- | 0.7 | -- | -- | -- |
Table 3: Clinical Outcomes
| CASE 1 | CASE 2 | CASE 3 | CASE 4 | |||||
|---|---|---|---|---|---|---|---|---|
| PRIOR TO SOTATERCEPT | AFTER DOSE 2 | PRIOR TO SOTATERCEPT | AFTER DOSE 2 | PRIOR TO SOTATERCEPT | AFTER DOSE 2 | PRIOR TO SOTATERCEPT | AFTER DOSE 3 | |
| 6 MWT (m) | 610 | 637 | 583 | 595.5 | 329 | 410 | 477 | 472 |
| ECHO | TAPSE: 1.17 | TAPSE: 1.44 | TAPSE: 1.03 | TAPSE: 1.35 | TAPSE: 2.3 | TAPSE: 2.7 | TAPSE:2.6 | TAPSE: 2.3 |
| TR: MILD | TR: MILD | TR: MILD- | TR: MILD- | TR 5.4 | TR 5.0 | TR 5.5 | TR 4.3 | |
| EST RVP: 80 | EST RVP: 100 | MOD | MOD. | EST RVP 115 | EST RVP 98 | EST RVP 119 | EST RVP 74 | |
| EST RVP:NO | EST RVP: 80 | |||||||
| EST | ||||||||
| WHO FC | 2 | 2 | 3 | 2 | 3 | 3 | 3 | 3 |
| IMPROVEMENT IN | --- | NONE | --- | Reports | --- | Reports | No | |
| SYMPTOMS | improved activity | improvement in SOB and | appreciable change in | |||||
| tolerance. | activity | symptoms | ||||||
| tolerance | ||||||||
21 Use of Pulmonary Vasodilators in Pediatric Patients With Congenital Heart Disease Awaiting Heart Transplantation
Carlos Sisniega, Juan Alejos
Division of Pediatric Cardiology, UCLA Mattel Children's Hospital, Los Angeles, California, USA
Patients with single ventricle (SV) physiology form a small subset of congenital heart disease (CHD) cases. Despite improved survival, complications like pulmonary vascular disease (PVD) affect outcomes. Pulmonary vasodilators (PV) are used sporadically but don't consistently reduce morbidity or mortality. This study evaluates the use of PV on heart transplantation (HT) candidates.
Retrospective cohort study of children < 18 years of age who were diagnosed with SV physiology who were awaiting HT due to Fontan failure between 2010 to 2022. Clinical, echocardiographic, and hemodynamic data were collected.
31 patients with were listed and had HT. 16 (52%) patients had SV physiology and 15 (48%) had biventricular physiology. Of the SV patients, 11 had subsystemic right ventricle and 5 had subsystemic left ventricle. Mortality was 6.4% at 28 days after HT and 32% thereafter. Pre-transplant hemodynamics showed a mean pulmonary artery pressure of 19 mmHg (IQR 17-22 mmHg), pulmonary artery wedge pressure of 15 mmHg (IQR, 12- 15), and PVRi 2.6 WUi (IQR, 2.6 (1.5-4.5). A total of 9 (29%) patients were placed on PV with use being highest at the pretransplant catheterization. 3 (10%) patients were placed on dual therapy with a phosphodiesterase inhibitor and an endothelin receptor antagonist. At 1 year following HT, only 2 patients were on PV. One year hemodynamics did not differ between patient who were on PV vs. patients naïve to PV.
There is a low prevalence of suspected PVD in the peritranplant period in pediatric patients with CHD awaiting transplant. PV use was minimal and mostly in the peritransplant period. There was no difference in hemodynamics when comparing those who used PV vs. naïve patients up to 1 year following HT despite PV wean. Weaning off PV following HT did not affect hemodynamics based on catheterizations within 1 year of HT.
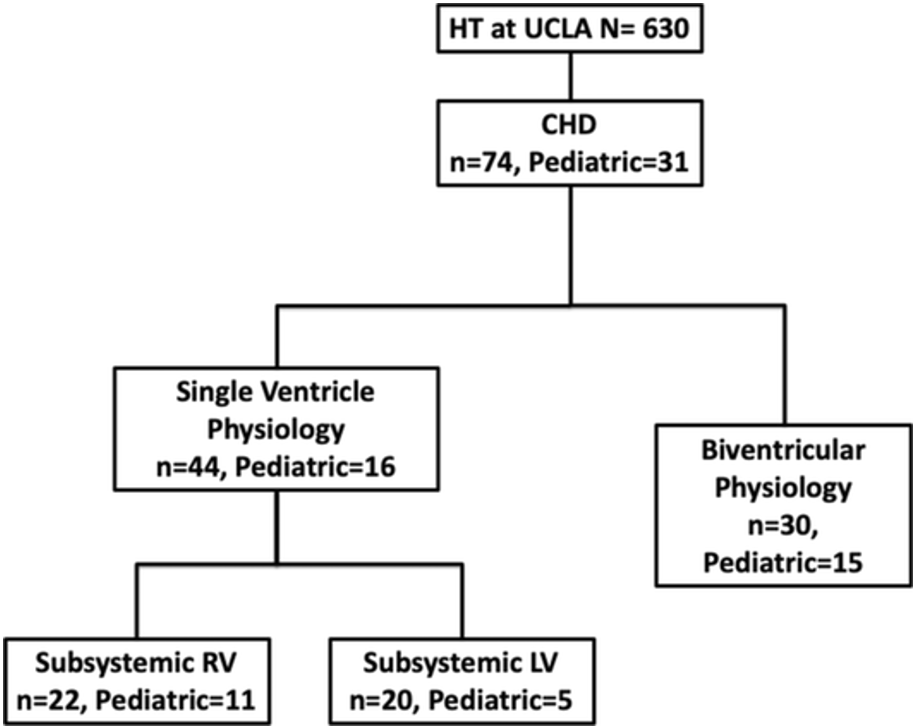
Figure 1.
| Age at transplant, years (IQR) | 1.4 (0.3-10.2) |
| Female, n (%) | 10 (32.3) |
| weight, kg (IQR) | 10.0 (5.0-32.6) |
| Single Ventricle | 16 (52.6) |
| Subsystemic ventricle | |
| Right, n (%) | 11 (69) |
| Left, n (%) | 5 (31) |
| Stage 1 SV repair | 15 (52) |
| BCPC, n (%) | 10 (34) |
| TCPC, n (%) | 4 (14) |
| Early Death (< 28 days post transplant), n (%) | 2 (6.4) |
| Late Death, n (%) | 9 (32) |
Table 1. Patient Characteristics
| Hemodynamics | |
| sPAP, median (IQR) | 21.5 (19.5-28) |
| dPAP, median (IQR) | 17.5 (16-1.5) |
| mPAP, median (IQR) | 19 (17-22) |
| PVRi, median (IQR) | 2.6 (1.5-4.5) |
| SVRi, median (IQR) | 9.4 (6.6-21.2) |
| PVR/SVR, median (IQR) | 0.2 (0.1-0.3) |
| PWAP, median (IQR) | 15 (12-15) |
| TPG, median (IQR) | 7 (5-10) |
| MAP, median (IQR) | 57 (52-62) |
| CI, median (IQR) | 4.65 (3.2-5.8) |
| Arterial saturation, median (IQR) | 80 (73-85) |
Table 2. Baseline Hemodynamics prior to heart transplant.
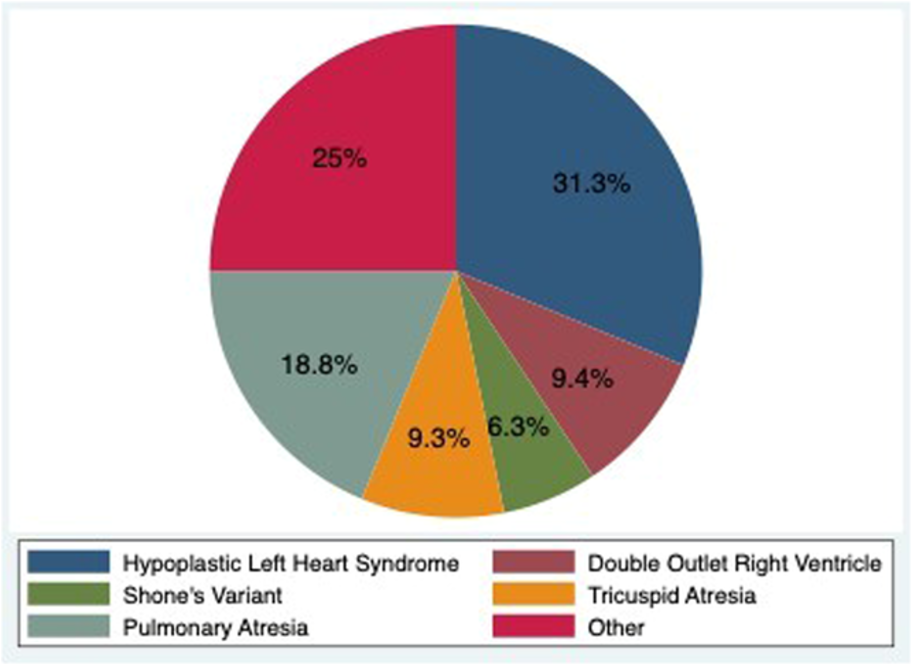
Figure 1. Distribution of Congenital Heart Disease.
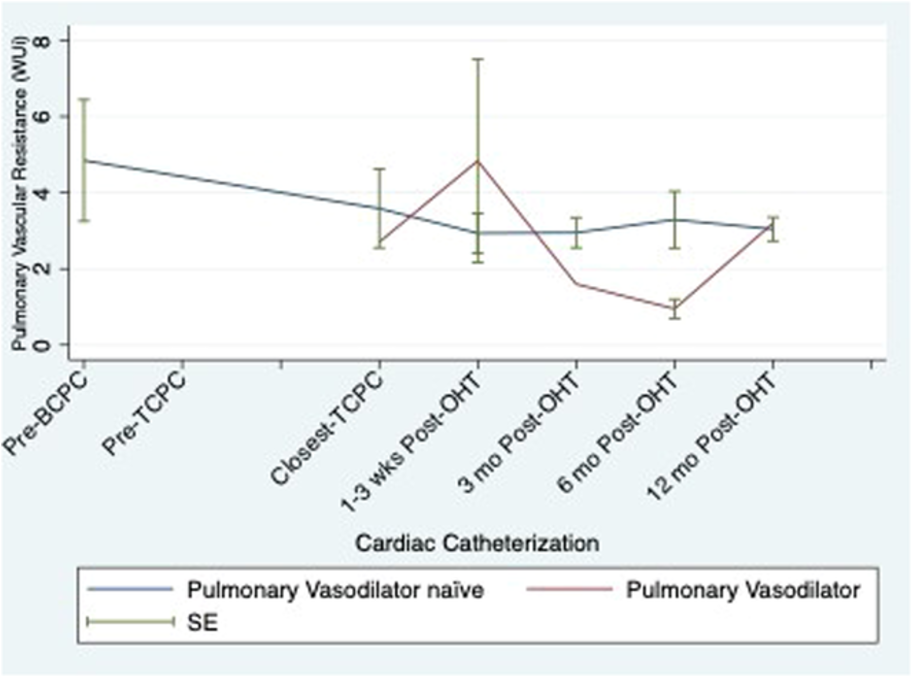
Figure 2. Serial pulmonary vascular resistance by use of pulmonary vasodilators.
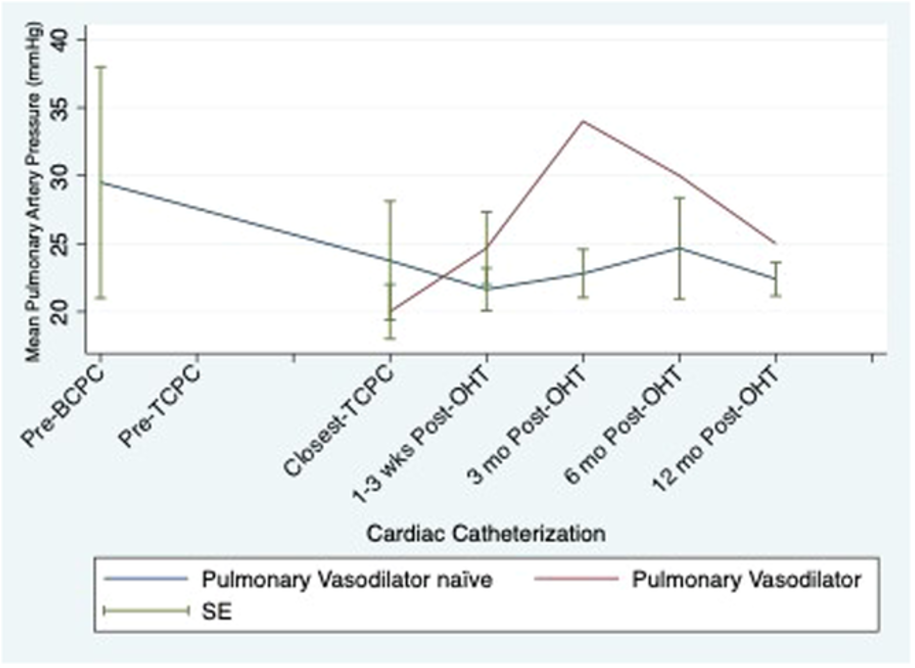
Figure 3. Serial mean pulmonary artery pressure by use of pulmonary vasodilators.
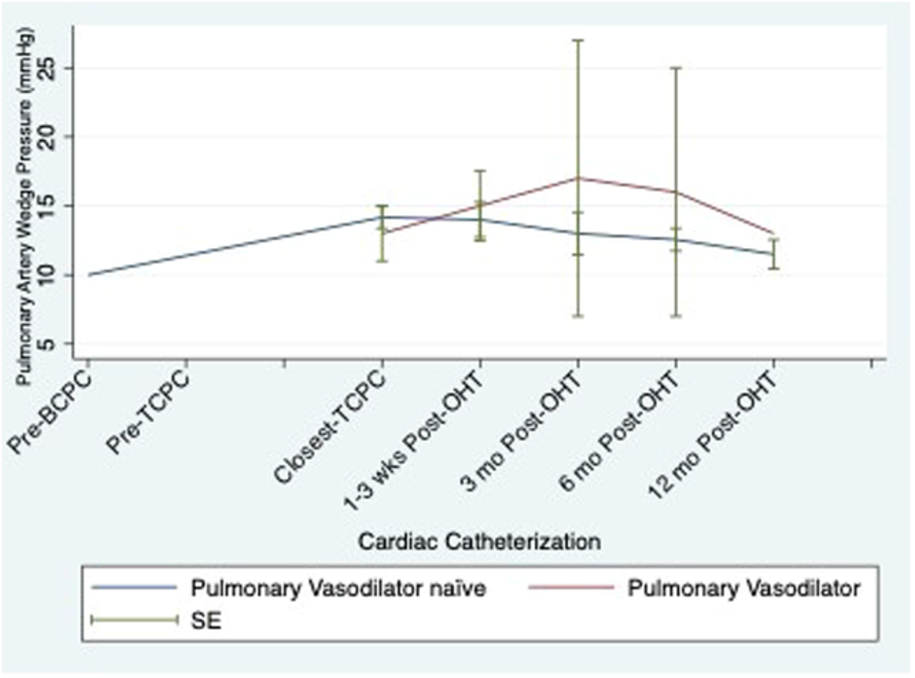
Figure 4. Serial pulmonary artery wedge pressure by use of pulmonary vasodilators.
| PVD medication | Pre-BCPC | Pre-TCPC | pre-OHT | 1-3 wks post | 3 mo post | 6 mo post | 12 mo post |
| PDEi | 1 | 1 | 9 | 6 | 3 | 2 | 2 |
| Sildenafil | 1 | 1 | 7 | 4 | 2 | 1 | 1 |
| Tadaladfil | 0 | 0 | 2 | 2 | 1 | 1 | 1 |
| ERA | 1 | 2 | 3 | 3 | 0 | 0 | 0 |
| Bosentan | 1 | 2 | 2 | 2 | 0 | 0 | 0 |
| Macitentan | 0 | 1 | 1 | 1 | 0 | 0 | 0 |
| DualTherapy | 0 | 1 | 3 | 3 | 0 | 0 | 0 |
Table 3. Frequency of pulmonary vasodilators throughout transplant course.
22 Clinical Pharmacist Role and Responsibilities Within Pediatric Pulmonary Hypertension Services
Meredith O'Neil, Neelam Bhatt, Megan Greene, Paul Critser, Nidhy Varghese, Stephanie Handler
Cincinnati Children's Hospital Medical Center, Cincinnati, OH, USA; Texas Children's Hospital, Houston, TX, USA; Children's Hospital Colorado, Aurora, CO, USA; Children's Wisconsin, Milwaukee, WI, USA
Medication regimens used for the management of pediatric pulmonary hypertension (PPH) are complex and high-risk requiring specialized expertise. Clinical pharmacists are essential in providing safe and effective care. However, there is no standardization of their role across institutions. Accreditation criteria for a Pulmonary Hypertension Association Center of Comprehensive Care (CCC) specifies necessity of a pharmacy with access to and proficiency with parenteral prostacyclin agents. However, the role and responsibilities of a clinical pharmacist are not well delineated. We aim to describe the current extent of involvement of clinical pharmacists within PPH services across the United States and identify disparities therein.
A survey was distributed to a pediatric cardiology pharmacist listserv with over 150 pharmacists to gather information on their various tasks and responsibilities in PPH management.
Survey responses from 27 pharmacists (24 institutions) were included. Overall, seventy percent of pharmacists reported < 10% of their FTE is utilized for PPH, and most pharmacists (23/27) reported primarily inpatient roles. Ninety-six percent (26/27) reported involvement in direct patient care (Figure 1).
Figure 1: Direct Patient Care Responsibilities
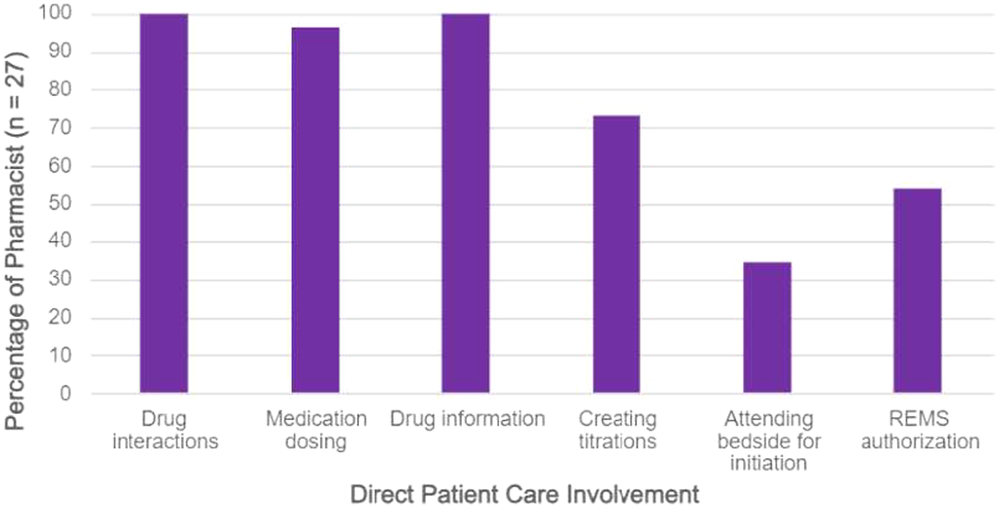
Pharmacists were actively involved in PH-related education (59% patient medication education and 82% healthcare professional education). Most respondents (93%, 25/27) did not have an established collaborative practice agreement allowing independent prescribing and only four reported consistent involvement in periodic review of high-risk patients (15%, 4/27). There was little involvement with specialty pharmacy coordination, prior authorizations, or PH scholarly activities (74%, 70%, 78%, reported no involvement, respectively). Notably, the extent of pharmacist involvement was higher in institutions designated as a CCC (Figure 2).
Figure 2: Percent of Institutions Reporting Pharmacists Role & Scholarly Involvement
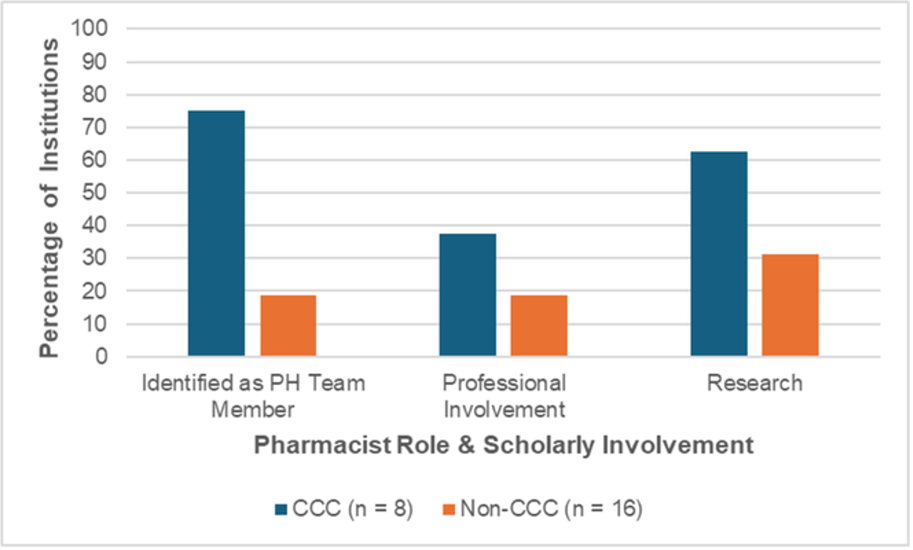
The extent of clinical pharmacist involvement on PPH services varies considerably across centers. Despite lack of dedicated time, pharmacists serve as important assets to the PPH service. More guidance from accrediting bodies describing specific roles and responsibilities for clinical pharmacists will assist in expanding dedicated pharmacy support in PPH.
23 Resolution of Pulmonary Hypertension in Severe Bronchopulmonary Dysplasia Without Pulmonary Vasodilator Use: A Validation Cohort
Meredith Riker, Hannah Cody, Emma Jackson, Stephanie Handley, Laurie Eldredge, Delphine Yung
Seattle Children's Hospital and University of Washington, Seattle, WA, USA
The protocol for treating BPD-PH at Seattle Children's Hospital optimizes respiratory support, treats potential aspiration and closes PDA and ASD before initiation of PH medication. We now report the outcomes of our second 3-year cohort.
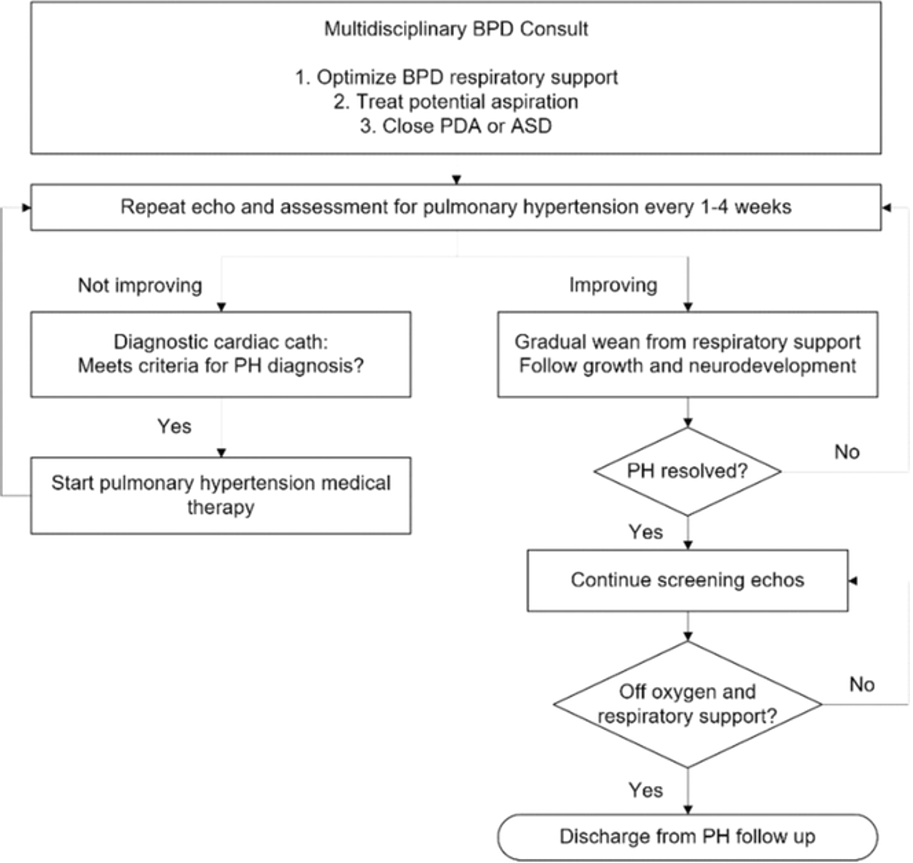
A retrospective review (2020–2023) of infants with BPD-PH analyzed demographics, comorbidities, treatments, PDA/ASD closure, and outcomes, including time to resolution. PH was defined by septal flattening, TR > 2.5 m/s, and low PDA/VSD gradient on echocardiogram.
79 patients had BPD-PH without critical congenital heart disease (Table 1). PH was first diagnosed at median 28 weeks PMA, resolving at median 39 weeks PMA, but recurred in 35 patients (46%) at median 47 weeks PMA. Most recurrences (n=31, 89%) resolved again by median 59 weeks PMA. A second recurrence occurred in 10 patients (13%).
Most initial and recurrent PH cases were linked to aspiration, pulmonary vein stenosis (PVS), or inadequate respiratory support. Second recurrences were due to aspiration (n=4), PVS (n=3), inadequate support (n=2), and other (n=1).
There were seven deaths, all unrelated to PH. PDA device closure was performed in 32 patients at median 31.9 weeks PMA, and ASD device closure in 8 patients at median 46 weeks PMA.
Three (4%) patients received sildenafil; two discontinued on arrival from outside hospitals, and one stopped after 11 days due to desaturation.

There appear to be two peak times of risk for PH: before 40 and after 47 weeks PMA. The first peak may be related to presence of PDA resolving after closure, while the second may stem from ASD, aspiration or respiratory support wean. Ongoing PH may indicate development of PVS. PH screening should continue until patients are off respiratory support and on full oral feeds. Despite a 9% mortality rate, no deaths were PH-related. Pulmonary vasodilator use may mask risk factors, prolonging lung disease.
24 Relationship Between End Systolic Eccentricity Index and Pulmonary Hypertension in Neonates With Congenital Diaphragmatic Hernia
Michael Cookson, Charles Simpkin, Mairead Dillon, MB, Jason Gien, Benjamin Frank
Section of Neonatology, Cardiology, Dept. Of Pediatrics, University of Colorado, Aurora, CO, USA
Pulmonary hypertension (PH) is common in infants born with congenital diaphragmatic hernia (CDH). Right heart catheterization (RHC) is the gold standard for diagnosing and monitoring PH but is limited in neonates due to clinical instability as well as anesthesia and procedural risks. Flattening of the ventricular septum is often present in PH and can be quantified with the eccentricity index (EI) during transthoracic echocardiography (TTE). EI has been validated in pediatric populations but there is limited evidence on the use of EI in patients with CDH. Unique to CDH patients is left heart structure hypoplasia which may impact assessment of EI. We sought to understand the relationship between EI and invasive metrics of PH by RHC in infants with CDH.
This was a single-center, retrospective cohort study of infants with CDH without complex congenital heart disease who had complete TTE data within 21 days of RHC between 2010-2024. Septal position at end systole and during max deformation was quantified by eccentricity index (EI) as EIs and EIm, respectively. EI was calculated as the ratio of left ventricle diameter parallel to the septum divided by the diameter perpendicular to the septum. Spearman's rank correlation was used to assess associations between TTE and RHC parameters.
Twenty-one patients were included in the analysis of which 90% had liver herniation at the time of CHD repair and 90% had left sided defects. The median age at time of RHC was 95 days (IQR, 52-159) and median time between ECHO and RHC was 6 days (IQR, 3-8). All patients met the diagnosis of PH with mean pulmonary artery pressures (mPAP) greater than 20 mmHg. EIs positively correlated with both mPAP and systolic pulmonary artery pressure (sPAP) with r= 0.5(CI 0.08-0.77) and 0.59(CI 0.21-0.82, respectively. EIm was not significantly correlated with mPAP but had moderate correlation, r=0.46 (CI 0.04-0.75), with sPAP. In a sub analysis, patients with a PDA (n=10) EIs did not correlate with sPAP, but had modest correlation with sPAP, r=0.67 (CI 0.12-0.91), in patients without a PDA (n=11).
EIs had moderate correlation with both mPAP and sPAP. EIs may prove to be a useful tool in trending CDH PH noninvasively while informing appropriate selection of infants requiring RHC, though sensitivity of the EIs may be affected by the presence of a PDA.
Inhaled treprostinil in mechanically ventilated children.
Diane Paulus, Ashley Hattie, Beth Malehorn, Robert Gajarski, Sarah Cohen
Nationwide Children's Hospital, Columbus, OH, USA
Pulmonary hypertension is attributable to lung disease in up to half of affected children, many of whom may require mechanical ventilation. For patients with heterogeneous lung disease, systemic pulmonary vasodilators may worsen ventilation-perfusion matching. This can be mitigated by administration of pulmonary vasodilators via inhalation. Inhaled iloprost has been successfully used in mechanically ventilated children with pulmonary hypertension, but its short half-life makes it difficult to use in the outpatient setting. Inhaled treprostinil (IT) is dosed less frequently and has been successfully used in children with pulmonary hypertension (PH), and so may be an attractive alternative agent in mechanically ventilated children.
A single-center retrospective chart review of all patients who received IT while mechanically ventilated.
53 children (53% female) at our center have received IT while mechanically ventilated. Most were infants, whose initial dose of IT was during their birth hospitalization.
Diagnoses included persistent pulmonary hypertension of the newborn (including congenital diaphragmatic hernia), bronchopulmonary dysplasia-associated PH, and congenital heart disease-associated PH. IT was administered through both endotracheal and tracheostomy tubes. Four children who were tracheostomy and ventilator dependent received IT for an average of four years, as part of their outpatient pulmonary hypertension regimen. IT was commonly used with other PH medications, including iNO (92%), sildenafil (38%), and bosentan (23%). In patients for whom there was an echocardiogram completed prior to and after the initiation of IT, without other interim interventions, there was echocardiographic evidence of improvement in right ventricular systolic pressure or function in most.
IT was discontinued due to adverse effects in nine (17%) (4 pulmonary hemorrhage, 4 hypotension, 1 oxygen desaturation). 38% received albuterol with doses of IT, though no bronchospasm was documented.
IT can be safely administered in mechanically ventilated children, is typically well tolerated, and appears to be effective.
25 Right Ventricular Strain Curve Patterns are Associated With Invasive Hemodynamics and Outcomes in Pediatric Pulmonary Arterial Hypertension
Karen Carvalho, Nazia Husain, Pei-Ni Jone, Amanda Hauck
Ann & Robert H Lurie Children's Hospital, Chicago, IL, USA
Post-systolic right ventricular free wall strain (RVFWS) curve patterns on echo correlate with RV diastolic function and disease severity in adults with pulmonary arterial hypertension (PAH). Clinical importance of RV strain patterns in children is unknown.
We aimed to describe RV strain curve morphology in children with PAH and assess relationships between patterns, hemodynamics and patient outcomes. We performed a retrospective cohort study of patients < 18yo with PAH. Demographic, echo and catheterization data at diagnosis and first follow-up catheterization were collected. Clinical composite endpoint was defined as need for septostomy, Potts shunt, lung transplant, or death. RVFWS was retrospectively performed for patients and 30 age- and sex-matched controls. Criteria for different strain curve patterns were identified. Differences in hemodynamics and outcomes between pattern groups were compared.
Thirty-two PAH patients were included with median clinical follow up of 5.6 years [range 0.1-15.5 years]. Three distinct strain patterns were identified (Figure 1a). Healthy controls exclusively demonstrated pattern 1. Peak RVFWS was lower in patients than healthy controls (19.3% [13.2%, 22.5%] vs 30.8% [28.5%, 33.0%] p< 0.01), but did not differ between patient groups. Mean PAP (35 [28-55], 40 [34-54], 62mmHg [48-76]; p=0.015) and PVR (5.5 [3.9-9.3], 8 [4.4-12.2], 16.1WUxm2 [12.4-25.5]; p< 0.01) increased from group 1 to 3 consistent with worsening disease severity.On 1 year follow-up echo, 21 (68%) had stable pattern 1 or improved pattern grade on therapy and none met endpoint. Among 10 patients with stable pattern ≥ 2 or pattern worsening, 70% met composite endpoint. Kaplan Meier analysis showed better transplant free survival in those with pattern 1 versus 2 or 3 at diagnosis (Figure 1b).
Distinct patterns of RV post-systolic strain morphology in pediatric PAH correlate with disease severity and outcomes. Further studies should evaluate their utility as a prognostic tool.
Figure 1
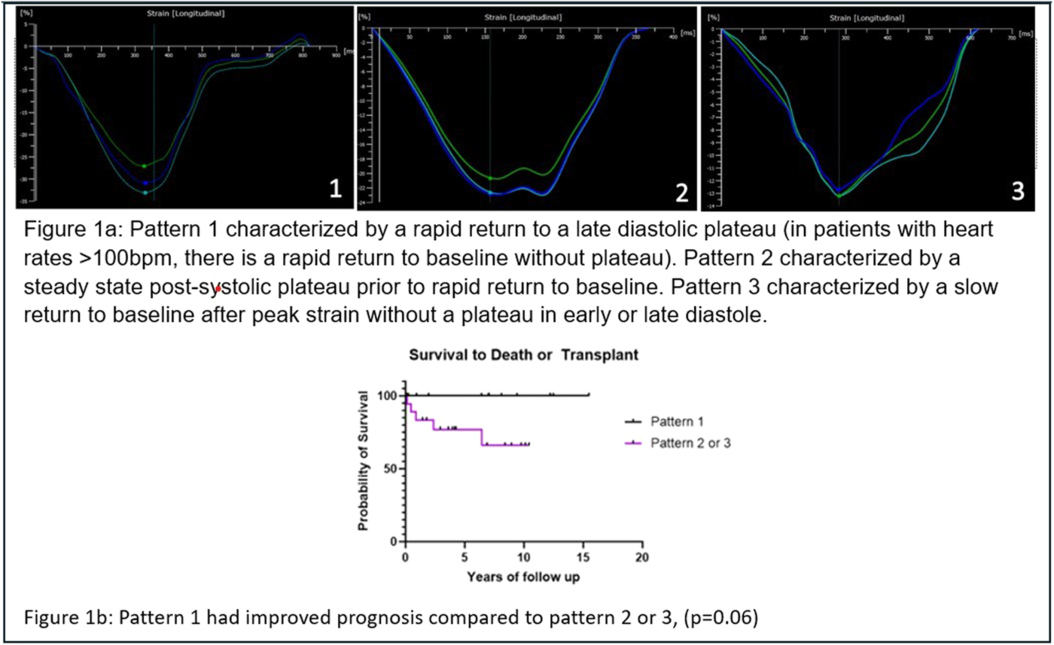
26 Hemodynamic Risk Factors for Death or Lung Transplantation in the TOPP-2 Registry
Usama Kanaan, Dunbar Ivy, Rolf Berger, Tilman Humpl, Damien Bonnet, David Bowers, Sonja Mariotti Nesurini, Maurice Beghetti M on behalf of the TOPP-2 Registry.
Emory University, Atlanta, Georgia, USA; Children's Hospital Colorado, Aurora, Colorado, USA; Beatrix Children's Hospital, Groningen, the Netherlands; The Hospital for Sick Children University of Toronto, Toronto, Ontario, Canada; M3C-Necker Hospital for Sick Children, Paris, France; University of Suffolk, Ipswich, United Kingdom; nspm ltd, Meggen, Switzerland; Children's University Hospital and University of Geneva, Geneva, Switzerland.
Right heart catheterization (RHC) remains the gold standard for diagnosis of pulmonary arterial hypertension (PAH) and is also recommended to monitor disease severity during follow-up. The prognostic significance of RHC-derived parameters in pediatric PAH remains unclear.
Tracking Outcomes and Practice in Pediatric Pulmonary Hypertension-2 (TOPP-2; NCT02610660) is an international, multi-center registry of incident pediatric PAH patients. Initial RHC diagnostic was required for inclusion. Follow-up RHC (FURHC) was performed in a subset of patients at treating physician discretion. Lung transplant- free survival was assessed by Kaplan-Meier analysis for candidate hemodynamic parameters at diagnostic RHC and FURHCs.
Thirty-one participating centers reported 983 individual catheterizations in 494 patients. Of these, 331 subjects met inclusion criteria for this sub-analysis based on PAH etiology (idiopathic n=182, heritable n=51, drug/toxin-induced n=3, or associated with congenital heart disease with incidental shunt n=50, repaired shunt n=41, or without prolonged initial shunt n=4). 178 of 331 patients (53.8%) had at least one FURHC.
Mean pulmonary artery pressure (MPAP) < 40 mmHg at initial RHC trended toward improved survival (p=0.052); however, achieving vs not achieving MPAP < 40 mmHg at first FURHC was strongly associated with freedom from death/transplant over the study observation period (mean observation time 70.7 vs 68.2 months with 96.5% vs 73.1% event-free; log-rank test p< 0.001). Pulmonary vascular resistance < 6.3 WU·m2 at diagnosis and first FURHC trended toward improved outcome (p=0.128 and 0.056, respectively), while cardiac index > 2.5 L/min·m2 did not predict significantly improved survival. Reducing MPAP between initial RHC and first FURHC without achieving the target threshold of 40 mmHg was not predictive of freedom from death/transplant.
In pediatric PAH, achieving MPAP < 40 mmHg at first FURHC is a strong predictor of survival. Future analyses should refine threshold values for MPAP and other hemodynamic parameters to enhance risk stratification and treatment strategies.

