Emergence of bullous pemphigoid under treatment of mycosis fungoides with mogamulizumab
Abstract
Mycosis fungoides (MF) is the most common cutaneous T cell lymphoma. Since 2018, mogamulizumab, an antibody directed to to the chemokine receptor CCR4, is licensed for the treatment of MF. Treatment with mogamulizumab is associated with the precipiation of different types of skin rashes summarized as mogamulizumab-associated rash. Here, we report the emergence of severe bullous pemphigoid (BP) in a patient suffering from severe MF and treated with mogamulizumab. BP is an autoimmune blistering skin disease causing severe pruritus and subepidermal blisters and, consequently, erosions. It is driven by autoantibodies against BP180, a protein of the dermal-epidermal adhesion complex. The parallel occurrence of MF and BP led to a bizarre clinical and histopathological presentation blending features of MF and BP. Among others, the patient developed multiple large erosions with a diameter of up to 15 cm, and histopathology featured subepidermal clefts with a mixed dermal infiltrate and atypical lymphocytes forming a superficial dermal lichenoid infiltrate and showing epidermotropism, including above the subepidermal clefts. Immunopathology revealing linear depositions of IgG and C3 at the dermal-epidermal junction and very high serum levels of anti-BP180-NC16A IgG were instrumental to diagnose BP and to distinguish it from mycosis fungoides bullosa, an extremely rare variant of MF. This case illustrates that immunopathology for BP should be conducted in patients with MF developing pruritus and blisters, although both can also be a symptom of MF. Our case alone does not allow determining whether the emergence of BP under mogamulizumab treatment was a mere coincidence or was in a causal relationship. The latter scenario would at BP to the possible clinical presentations of mogamulizumab-associated rashes.
INTRODUCTION
Mogamulizumab is a defucosylated, humanized, monoclonal IgG1 κ antibody directed to the cc chemokine receptor 4 (CCR4).1, 2 Binding of mogamulizumab to CCR4 inhibits CCR4 signaling and initiates the depletion of CCR4+ cells, specifically skin-homing malignant T cells, T helper (TH)2, and regulatory T cells (Tregs).3 Mogamulizumab was developed for the treatment of the Sézary syndrome and mycosis fungoides (MF).4
MF is the most frequent cutaneous T cell lymphoma and mostly exhibits a mild and very slowly progressing course of disease.5 Mogamulizumab is approved for use in MF patient having received at least one prior systemic therapeutic.2 The most common side-effect of mogamulizumab are skin rashes. So, the term “mogamulizumab-associated rash” (MAR) was coined.3 These rashes are heterogeneous in their morphology. They predominantly present as folliculotropic MF–like scalp plaques with alopecia, papules and plaques or photoaccentuated, morbilliform or erythrodermic dermatitis.3
Bullous pemphigoid (BP) is an autoimmune blistering skin disease defined by the generation of autoantibodies against BP180, a protein of the dermal-epidermal adhesion complex.6 Upon binding at the dermal-epidermal junction, immune cells, particularly granulocytes are recruited into the dermis where they degrade the dermal-epidermal adhesion complex consequently leading to the formation of subepidermal clefts clinically presenting as blisters and finally erosions.6 In addition, BP patients typically suffer from strong pruritus,7 a symptom shared with MF.5 The treatments of BP and MF fundamentally differ. BP usually requires aggressive, long-term immunosupression to be controlled,7 whereas the treatment of MF aims at depleting tumor cells and preventing their proliferation,2 a strategy compromised by strong immunosuppression.
CASE REPORT
In a 72-year-old Caucasian male bullous pemphigoid (BP) manifested 20 months after the patient had been diagnosed with mycosis fungoides (MF). When BP emerged, the MF was at stage IIB, and the patient had received 10 cycles of 1 mg/kg mogamulizumab during the last 5 months combined with irradiation of tumor nodules at the left hand and upper arm. Previous treatments had included oral PUVA therapy for 2 months, bexarotene plus bath-PUVA therapy for 4 months, and pegylated interferon-α plus bath-PUVA for 7 months.
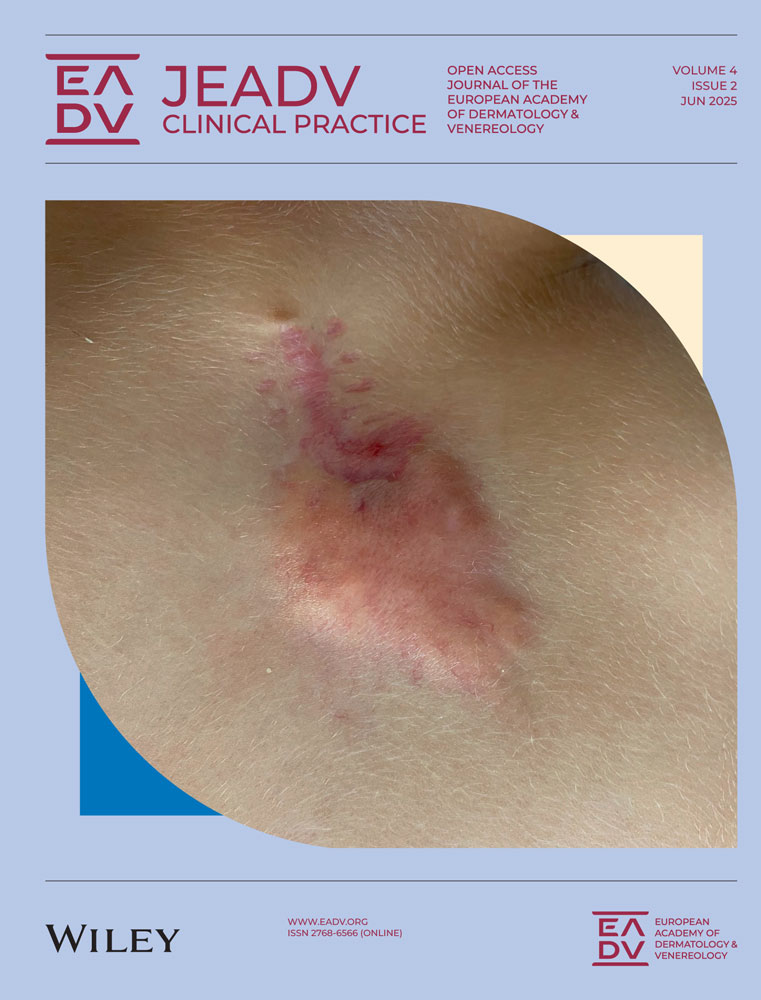
BP first manifested with blisters on the lower legs. Mogamulizumab was, consequently, discontinued, brentuximab vedotin (1.8 mg/kg) was given once, and 60 mg/day prednisolone were initiated. One month later, the patient was transferred to the University Hospital of Lübeck. By then, the blisters had turned into large erosions with a diameter of up to 15 cm affecting entire body except the face (Figure 1a). Direct immunofluorescence microscopy of perilesional skin uncovered IgG and C3 depositions at the dermal-epidermal junction (Figure 1b), and indirect immunofluorescence on salt-split skin detected circulating IgG and IgA autoantibodies binding to the epidermis. By ELISA, a high level of anti-BP180-NC16A IgG (1972 U/mL) was determined. Histopathology showed subepidermal clefts and mixed inflammatory dermal infiltrates (Figure 1c). Thus, the diagnostic criteria of bullous pemphigoid (BP) were met.8 Intriguingly, there was a blend of MF and BP features with skin lesions additionally infiltrated by atypical lymphocytes forming a superficial dermal lichenoid infiltrate and showing epidermotropism, including above the subepidermal clefts (Figure 1d).

BP was treated with topical clobetasol propionate 20 g twice daily from neck to toes, intravenous pulses of dexamethasone (100 mg/day for 3 consecutive days), and 125 mg/day dapsone. The latter was discontinued after 1 month due to anemia. Oral prednisolone was slowly tapered from 30 to 5 mg/day, and brentuximab vedotin was discontinued.
The response of BP to this treatment was slow but new skin lesions ceased emerging and existing erosions started epithelizing. However, the patient contracted atypical pneumonia which was treated with piperacillin/tazobactam and meropenem. To limit immunosuppression, dexamethasone was replaced by intravenous immunoglobulins (2 g/kg body weight) of which two cycles were finally applied.
Thus, BP was controlled but tumor nodules progressed although bexarotene (525 mg/day) and low-dose CHOP therapy (cyclophosphamide, doxorubicin, vincristin, prednisone) were (re-)initiated. Shortly after, the patient succumbed to lobar pneumonia.
DISCUSSION
Our case demonstrates that MF and BP can overlap leading to atypical skin lesions and an unusually severe disease. Notably, there is an extremely rare blistering variant of MF, MF bullosa, featuring a clinincal and histopathological presentation similar to the case reported here,9, 10 and low levels anti-BP180-NC16A IgG are also prevalent in about 1%–2% of healthy individual.11 The detection of linear depositions of IgG and C3 was therefore essential to distinguish BP from MF bullosa. The diagnosis BP is our patient is additionally supported by our patient's exceedingly high levels of anti-BP180-NC16A IgG.
Our case illustrates that immunopathology to diagnose BP should be considered in patients presenting with blisters and/or erosions under mogamulizumab. The emergence of MAR is usually associated with a good treatment response to mogamulizumab.12 In our case, however, such a pronounced and long immunosuppression was required to control BP that the patient succumbed to pneumonia.
The patient developed BP after 5 months on mogamulizumab. We cannot distinguish whether the emergence of BP under mogamulizumab was a coincidence or caused or promoted by mogamulizumab. CCL17/TARC, a ligand of CCR4, is increased in BP but its significance is unknown,13 and TH2 cells and Tregs have both been implicated in BP but their role is unknown.14, 15 Thus, it is possible that mogamulizumab may directly interfere into pathways involved in the pathogenesis of BP.
AUTHOR CONTRIBUTIONS
Lukas D. Uleer and Christian D. Sadik wrote the paper. Iakov Shimanovich and Jasper N. Pruessmann performed immunopathological and histopathological analyses. Lukas D. Uleer, Carmen Loquai, Iakov Shimanovich, Cyrus Khandanpour, Jasper N. Pruessmann, Patrick Terheyden, Christian D. Sadik analyzed the case and edited the manuscript.
ACKNOWLEDGMENTS
The authors have no funding to report.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
ETHICS STATEMENT
All patients in this manuscript have given written informed consent for participation in the study and the use of their deidentified, anonymized, aggregated data and their case details (including photographs) for publication. Ethical Approval: not applicable.
Open Research
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analysed in this study.