

Innovative methods in soil phosphorus research: A review
Abstract
Phosphorus (P) is an indispensable element for all life on Earth and, during the past decade, concerns about the future of its global supply have stimulated much research on soil P and method development. This review provides an overview of advanced state-of-the-art methods currently used in soil P research. These involve bulk and spatially resolved spectroscopic and spectrometric P speciation methods (1 and 2D NMR, IR, Raman, Q-TOF MS/MS, high resolution-MS, NanoSIMS, XRF, XPS, (µ)XAS) as well as methods for assessing soil P reactions (sorption isotherms, quantum-chemical modeling, microbial biomass P, enzymes activity, DGT, 33P isotopic exchange, 18O isotope ratios). Required experimental set-ups and the potentials and limitations of individual methods present a guide for the selection of most suitable methods or combinations.
1 Introduction
Serious concerns within the scientific community and general public about future global supplies of phosphorus (P) fertilizer (Edixhoven et al., 2013; Gilbert, 2009; Cordell and White, 2011; Obersteiner et al., 2013; Heckenmüller et al., 2014) have initiated novel research platforms (e.g., the European Sustainable Phosphorus Platform, the US Sustainable Phosphorus Research Coordination Network, the Leibniz ScienceCampus Rostock “Phosphorus Research”). Improvements in the understanding and management of the soil P cycle depend on advances in analytical and theoretical methods of investigation. Phosphorus exhibits a great diversity of inorganic P (Pi) and organic P (Po) species, with some of them occurring in trace amounts in complex environmental matrices. Many of the species are involved in abiotic and biotic reactions; these differ widely and remain somewhat unresolved, making the elucidation of the agricultural and environmental P cycles a particularly challenging research topic.
Advances in understanding and elucidating these cycles inevitably require a toolbox of appropriate methods that can tackle the following urgent questions: (1) how much total P (Pt), Pi or Po is in the soil system?; (2) which P species or molecules are abundant at what concentrations?; (3) where in the soil do these P species or molecules exist, i.e., in which soil compartments or at which reactive surfaces?; (4) in which reactions are these species involved when under the influence of soil management or environmental changes? Often these topics are interlinked; for instance, process understanding (4) is deduced from changes in the P species and pools (2) that occur at specific spatial scales or locations (3). These multi-scale interactions among P forms and processes strongly call for the application of innovative methods which go beyond operationally defined P pools obtained from established soil P tests and fractionation methods. Methods or method packages are needed which also access P structural information both across spatial and temporal scales; this will enable a critical shift in our understanding from operationally to structurally and functionally-defined fractions (Condron and Newman, 2011).
This review is based on the discussions and contributions at the international workshop “Innovative methods in soil phosphorus research” on September 27, 2012, hosted by the recently established multidisciplinary Leibniz-ScienceCampus “Phosphorus Research Rostock” at Rostock University. The aim is to provide a critical review of state-of-the-art techniques rather than an all-encompassing publication of all analytical techniques for soil P. General overviews and comprehensive compilations of methods in environmental P research are already published (e.g., Sparks et al., 1996). Here we provide a concise and critical overview of state-of-the-art methods in environmental P research, which identifies the potentials and limitations of currently available analytical methods.
2 The soil P cycle in a nutshell
A conventional, simplified scheme to display the basic pools and processes in the soil P cycle involves (1) the inputs (I1 to I3), (2) the pools (P1 to P5) and reactions (R1 to R4) in soil, and (3) the P losses from soil (L1 to L3) (Fig. 1). Initially, P enters the soil through weathering of P containing minerals (mostly apatites: Ca10X(PO4)6, where X = F−, Cl−, OH− or CO32-) from parent rock (concentrations < 10−2 g P kg−1) (I1). The major P input to agricultural soils originates from manure, agricultural and municipal byproducts, and from mineral fertilizers (10−1 to 101 g m−2 y−1). Soils in natural and managed ecosystems also receive P from litter and residues of the vegetation and remnants of animals (I2). Other P inputs derive from atmospheric sources (wet and dry deposition: 10−2 to 100 g m−2 y−1) (Tipping et al., 2014) (I3). Globally, P inputs are distributed very unevenly; soils in less developed countries with low-input farming systems are undersupplied and those in industrialized countries often over supplied. Moreover, soils in intensively farmed Western European and North American areas may receive more P from atmospheric deposition (up to 10 kg ha−1 y−1; Tipping et al., 2014) than agricultural soils in African small-scale farming systems receive fertilizer- and manure P (< 5 kg ha−1 y−1; Chianu et al., 2012).
Total P concentrations in soils (sum of P1 to P5) range from 101 to 103 g P kg−1, depending on soil horizon (subsoil < topsoil), substrate (sandy < loamy), pedogenesis (older < younger), land use (forest < pasture < agriculture) and its land-use intensity (extensive < intensive). In soil the P is bound and cycling in various chemical compounds that differ in their composition (simplified as P pools in Fig. 1), their reactions with the liquid and the solid soil phase, and in their availability to microorganisms and crops; these components and processes are evident in the large central part in Fig. 1. Since a direct chemical-analytical speciation of all soil P compounds is not yet possible, the description and visualization of the total soil P involves chemically or mineralogically defined P compounds as well as operationally defined pools of P compounds with similar turnover rates (slow vs. rapid). These operationally defined P pools are measured by sequential fractionation schemes (e.g., Hedley et al., 1982); these schemes were reviewed recently (Negassa and Leinweber, 2009; Condron and Newman, 2011) and, therefore, are not included in the present review.
Generally, inorganic (Pi) and organic (Po) compounds can be distinguished analytically by two separate P determinations: in soil solution samples through a photometric phosphate determination (Pi) and an atomic emission spectrometric determination with inductively coupled plasma (ICP-OES = Pt; Po = Pt – Pi), and in soil solid samples by Pt (oxidation + digestion) and Pi (digestion) determinations or by more sophisticated analytical methods—as described in the following sections. The Pi/Po ratios in soil solid samples range from 0.1 to 3, depending on pedogenesis and organic matter (OM) content (older soils < younger soils, organic soils < mineral soils).
Defined Pi-containing minerals include the various Ca-phosphates, such as Ca(H2PO4)2 · H2O (monocalcium phosphate), CaHPO4 · 2H2O (dicalcium phosphate dihydrate = brushite), CaHPO4 (dicalcium phosphate = monetite), Ca8H2(PO4)6 · 5 H2O (octacalcium phosphate), Ca5(PO4)3OH (hydroxyapatite), and Ca5(PO4)3F (fluoroapatite). Other defined Pi minerals in soil are variscite (AlPO4 · 2 H2O), strengite (FePO4 · 2 H2O), and vivianite [(Fe3(PO4)2 · 8 H2O)]. Together, these Ca-phosphates contribute to I1 and P5 in Fig. 1. Their solubilities and, therefore, potential contributions to soil solution P concentration (R4) and speciation are different, and depend on pH and ion activities in solution. The Ca-phosphates are generally less soluble (= more stable) at high pH and the Al- and Fe-phosphates less soluble at low pH. In addition to precipitation from the soil solution (R4), portions of these phases can originate directly from the inputs (especially weathering of P minerals and P fertilizers).
Despite its relatively small contribution (≈ 10−1%) to the Pt pool, soil solution P (P2) is of critical importance; it acts as the reservoir for the P uptake by plants (L3) and microorganisms (R3), and is central to all sorption/desorption-, precipitation/dissolution-, and immobilization/mineralization reactions as well as leaching and runoff losses. The P concentrations in soil solution can vary widely, from 10−3 to 101 mg L−1, depending on Pt content, dominant P compounds, and soil composition and properties. Field crops, however, require > 2 × 10−1 mg P L−1 for optimal growth. The most abundant Pi forms in soil solution are the anions H2PO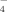 and HPO
and HPO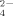 ; both can be taken up by plants and microorganisms. The ratio of their concentrations falls from 10 at pH 6 to 0.1 at pH 8; at pH > 9.5 the ion PO
; both can be taken up by plants and microorganisms. The ratio of their concentrations falls from 10 at pH 6 to 0.1 at pH 8; at pH > 9.5 the ion PO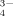 may occur in the soil solution. A substantial, yet highly variable proportion of total P in soil solution is Po; published data range from 20 to 90% (Adesanwo et al., 2013: fertilized and non - fertilized soils in Manitoba) and > 80 to 99% (Shand et al., 1994: P poor Cambisols in Scotland). It appears that the contribution of Po to soil solution P increases with decreasing total P concentration, but published data are surprisingly sparse. This issue requires more research since significant proportions of Po in soil leachates seem to be plant available (McDowell and Koopmans, 2006).
may occur in the soil solution. A substantial, yet highly variable proportion of total P in soil solution is Po; published data range from 20 to 90% (Adesanwo et al., 2013: fertilized and non - fertilized soils in Manitoba) and > 80 to 99% (Shand et al., 1994: P poor Cambisols in Scotland). It appears that the contribution of Po to soil solution P increases with decreasing total P concentration, but published data are surprisingly sparse. This issue requires more research since significant proportions of Po in soil leachates seem to be plant available (McDowell and Koopmans, 2006).
The Pi solubility and Pi removal from the soil solution depend on soil mineralogy and pH. In alkaline soils, the P concentration in soil solution gradually decreases through precipitation of less soluble Ca-phosphates (R4). In highly weathered acidic soils rich in Al- and Fe-oxides (e.g., Ferralsols that predominate in subtropical and tropical regions), Pi is strongly sorbed on to the edges of silicate clay minerals and to pedogenic Al- and Fe-oxides (R1). Over time the Pi sorption may become gradually stronger by a slow diffusion of phosphate into micropores forming “occluded P” or even transitions to precipitated Al- and Fe-phosphates (P1 to P5). This process, contributing to the low efficiency of fertilizer P in low pH soils, may be partly avoided or reversed by low molecular organic anions and higher molecular organics such as fulvic and humic acids that compete with phosphate for positively charged binding sites. Chelating organic compounds also may contribute to the phosphate desorption. However, this commonly held view on the role of organic molecules was questioned by Guppy et al. (2005); these workers criticized a number of studies for not considering the P release from OM, thereby overlooking the effect of elevated P concentrations in equilibrium solution. There is substantial evidence that redox dynamics are closely linked to those of sorbed and dissolved phosphate because of the direct influence of redox on the solubility of Al- and Fe-oxides.
The speciation of sorbed, precipitated and otherwise mineral-bound P in the solid phase is further complicated by the ubiquitous presence Po and, as such, continues to present a major analytical challenge (cf. chapter 3). In short, the sorbed and precipitated Pi pools can be sequentially extracted by anion exchange resins (weakly sorbed P), NaHCO3 (stronger sorbed P and labile Al-, Fe-phosphates), NaOH (Al-, Fe-phosphates) and strong acids (H2SO2, HCl: Ca-phosphates). However, these extracts also commonly contain substantial amounts of Po.
The majority of Po is bound either in nonliving organic matter (P3), the majority of which may be humic substances (stable, slow turnover), or in the soil biomass, the latter displayed in Fig. 1 as P4 “Microbial biomass P” (Pmic; labile = fast turnover). Chemically, phytic acid salts (inositol hexaphosphate or IP6 salts) and plant assimilates predominate in the Po pool (> 50%). These Po phytates and other sugar phosphates occur in polymeric forms; thus, they can be bound by soil minerals and/or humic substances. Other Po compounds are nucleotide phosphates and phospholipids; these form substantial parts of the Pmic. Typically, the Pmic fraction accounts for 2 to 5% of Po in arable and up to 24% in grassland soils (Turner et al., 2001) and can be estimated using a well established fumigation/extraction technique that measures soil microbial biomass (Brookes et al., 1982; cf. chapter 4.3). The two Po pools (P3 and P4 in Fig. 1) interact with the soil solution by immobilization (P uptake and temporary binding in organic compounds) and mineralization (cell decay and enzymatic hydrolysis) processes (R2 and R3) at many different rates. Due to its rapid cycling, Pmic is considered very important for plant nutrition, whereas the cycling and ecological importance of Po in non-living OM have received much less attention. Finally, Pmic also is a potential source of Po in non-living OM if P containing microbial necromass becomes stabilized in humic substances; this pathway has scarcely been studied.
There are three major pathways of P loss from soil: (1) erosion, (2) leaching (both non-intended and sometimes mutually interlinked, and (3) the uptake by plants and removal with harvests. Erosive P losses at one site may result in P inputs at other sites. Typically, this leads to P-depleted upslope and summit positions in fields and P-enriched downslope and depression locations, the latter often close to vulnerable watercourses. The transfer of P from soil to water, both by erosive (runoff) and leaching processes and pathways, has received much attention because of the adverse effects on water quality (eutrophication). Phosphorus transfers can be expressed as a function of the dominant P forms, the mechanisms of their release from soil to water and the hydrological pathways of their transport (Leinweber et al., 2002). As such, P from all pools, shown in Fig. 1, can be transferred or lost but in different quantities and rates. Leaching losses are restricted largely to the soil solution P (dissolved and colloidal P), but particulate P can also be be lost especially in cracking clays with shallow tile drains (Addiscott et al., 2000). Under intensive agriculture, leaching losses reach critical levels when the P sorption capacity of the Al- and Fe-oxides is saturated by long-term P inputs to sandy soils that far exceed the plant uptake. Thus, non-intended P losses tend to increase when P inputs exceed projected crop P uptakes. Phosphorus removal with crop harvests is in the range of 2 to 5 × 100 g m−2 y−1, and ideally this rate should be added as manure or fertilizer inputs. A key research challenge for the future is to meet crop P requirements at lower external P inputs; this promises to radically reduce both undesired adverse P losses to waterways and our dependency on mineral P fertilizers. This challenge has stimulated significant progress in the development and application of methods in soil P research; these specialized advances are reviewed in the following chapters.

Schematic presentation of the soil phosphorus (P) cycle. The names in the black boxes (I = Inputs, P = Pools, R = Reactions, and L = Losses) are also used in Table 2 (below) to indicate which methods are suitable to characterize the respective part of the P cycle.
3 Spectroscopic and spectrometric P speciation methods
3.1 1D and 2D 1H–31P NMR Spectroscopy
Phosphorus has only one isotope, 31P, which is NMR-active with the highest sensitivity among the major elements cycling in soil (e.g., 13C and 15N). Since 31P is the only naturally occurring P isotope, all P atoms in an environmental sample are potentially accessible by NMR spectroscopy. Each P atom in any chemical structure gives rise to a specific 31P NMR signal; signal integrals are proportional to the quantity of the individual P species in the sample. The position of a 31P signal in an NMR spectrum—its chemical shift (in ppm) relative to an external standard, e.g., phosphoric acid or methylene diphosphonate (MDP)—is a measure of the electron density around a considered P atom. Tabulated chemical shifts of various environmentally relevant Po and Pi compounds are available (e.g., Cade-Menun, 2005; Turner et al., 2003a, b; Vestergren et al., 2012). Therefore, 31P NMR spectroscopy enables the quantitative identification of various P species in environmental samples.
31P NMR spectroscopy can be applied both to solid and liquid samples. Solid-state 31P NMR enables direct measurements of unaltered soil samples since neither extraction nor extensive pretreatments are required. The technique has been applied mostly to speciate Pi in environmental samples (e.g., Frossard et al., 1994; McDowell et al., 2002; Dougherty et al., 2005). Additionally, 31P NMR solid-state has been successfully applied to, e.g., investigate the mechanism of phosphate sorption on aluminum hydroxides under different environmental conditions (Li et al., 2013a) or kaolinite (Van Emmerik et al., 2007). However, more widespread application of solid-state NMR in soil science is still hampered by two main factors: the frequently low P concentration in soils and the poor spectral resolution. In particular, spectral overlaps hinder unambiguous signal assignments to individual P compounds and their subsequent quantification. The insufficient resolution originates from signal broadening due to fast relaxation times which, in soil, are accelerated further by paramagnetic metal cations such as Mn and Fe, and the heterogeneous nature of the sample (Cade-Menun, 2005). Furthermore, Dougherty et al. (2005) reported that interference from paramagnetic ions also hindered the quantitation of P by spin- counting solid-state NMR, whereby only a fraction of total P could be detected. Attempts to remove paramagnetics from soil samples to improve solid-state NMR spectra have yielded inconsistent results (e.g., McDowell and Smernik, 2010).
In order to obtain quantitative information, one-dimensional (1D) liquid-state or solution 31P NMR has been the method of choice to speciate Po in soil (Newman and Tate, 1980). The main advantage of solution- over solid-state 31P NMR is the generation of significantly higher spectral resolutions. The spectra are clearer and more distinct in their features, enabling more confident identification and quantification of P species of various classes such as orthophosphate, polyphosphate, pyrophosphates, diphosphate, phosphonates, monoesters, and diesters. A prerequisite for solution 31P NMR is the extraction of as much P as possible from the soil sample. Advantages and disadvantages of several extraction procedures and their effects on the resolution of 31P NMR spectra have been discussed extensively in the literature (e.g., Cade-Menun and Preston, 1996; Turner et al., 2005, Cade-Menun and Liu, 2014). Early research in solution-state 31P NMR used NaOH alone or a NaOH/NaF mixture to extract soil P; however, these chemicals lacked extraction efficiency and appeared to control the spectral composition (Amelung et al., 2001). One of the most common extraction protocols nowadays simply involves shaking the soil with 0.25 mol L−1 NaOH and 0.05 mol L−1 Na2EDTA for 16 h, followed by lyophilization. However, as with any alkaline treatment, there is a risk of sample alteration. This involves distortion of the original distribution of P compounds in the sample as a result of (1) selective extraction of certain P species, (2) compound-specific vulnerability to alkaline hydrolyses (especially diesters), and (3) possible mobilization, due to strongly alkaline conditions, of inavailable orthophosphate from soil P minerals (e.g., Ca-, Fe-, and Al- phosphates) and P complexed at surfaces, which would result in a overestimation of the soil orthophosphate pool. Nonetheless, despite these shortcomings, solution 31P NMR has been used to characterize Po in a wide variety of environmental samples, often correlating P speciation with bioavailability (Turner et al., 2003a, b; Ahlgren et al., 2005). However, the identification and quantification of individual P species, especially in the monoester and diester region of the spectra, remains a considerable challenge (Doolette et al., 2011; Makarov et al., 2002). A fundamental issue still hampering the use of solution 31P NMR in soil science is the variation in soil matrices, affecting the reproducibility of the chemical shifts. NaOH/EDTA mixtures co-extract paramagnetic Fe and Mn; however, in spite of complexation by the EDTA, these species remain major sources of line broadening in solution 31P NMR spectra (Cade-Menun et al., 2005; Newman and Tate, 1980). For many years, in order to enhance spectral resolution, the identification of specific P compounds in these extracts relied on spiking experiments (Doolette et al., 2009). However, while this approach allows the identification of spectrally distinguishable P species, it fails to differentiate between P compounds with very similar chemical shifts due to the spectral overlap. Therefore, quantification of individual P species via integration of their NMR resonances can be misleading and, as a consequence, concentrations of common soil Po species can be over- or underestimated (Doolette et al., 2009; Vestergren et al., 2012). In order to improve the capabilities of solution 31P NMR, several pre- and post-extraction treatments have been used to enrich P (enhance signal-to-noise ratio) and to eliminate the effects of paramagnetic metal ions (reduce line broadening) (Ding et al., 2010). A new promising protocol to reduce line broadening was recently reported by Vestergren et al. (2012), which is based on sulfide precipitation. In this approach, lyophilized NaOH/EDTA extracts are re-dissolved in D2O, and Na2S is added at 5 to 10 mol equivalents of the Fe concentration in the extract. This large excess of sulfide maintains reductive conditions, thereby preventing the formation of Fe-hydroxides which otherwise could remove P from solution; the sulfide step in the procedure maintains a P recovery as high as 94 to 105% (Vestergren et al., 2012). These workers also showed that this pretreatment significantly reduces line broadening, improves the spectral resolution, reduces resonance overlap, and, therefore, allows the identification and quantification of an increased number of P species. Nevertheless, more investigations are needed to test the efficacy of this prolonged method; e.g., in relation to the risk of increased hydrolysis of P diesters, and for various soil types and other environmental samples such as manure and sediments.
In spite of extensive research, 1D solution 31P NMR still fails to identify without ambiguity all P species in complex matrices such as soil extracts due to the vast number of P species present and their characteristically small chemical shift dispersion. One approach to overcome this limitation is by linking 31P signals to 1H signals in P–O–CH moieties by so-called two-dimensional (2D) liquid 31P, 1H HSQC (Heteronuclear Single Quantum Correlation) NMR spectroscopy. The obtained 2D NMR spectra comprise a 31P axis (vertical) and a 1H axis (horizontal). Generally, such spectra can be viewed as maps, where each “top” (cross peak) represents a single P species which is characterized by its 31P chemical shift and the 1H chemical shift in its P–O–CHn moiety. The main advantage of this 2D approach is the large amount of information obtained. While in 1D solution NMR P components can be separated only according to their 31P chemical shifts, the second dimension allows a correlation with the 1H chemical shifts of protons in the direct molecular vicinity of the P nucleus. Because the molecular structures in the vicinity of the P nucleus vary among P species, the 1H signals contain characteristic proton-proton fine structures. The combined information derived from 1H chemical shifts and fine structures allows clear separation and unambiguous identification of P species even if they have identical 31P NMR chemical shifts. In biochemistry, 2D 31P, 1H NMR methods are well established to identify phospholipids in complex mixtures (Petzold et al., 2009).
Initial applications of 2D 31P, 1H NMR in soil P-studies were made possible by the substantial reduction of line broadening by the aforementioned sulfide pretreatment (Vestergren et al., 2012). Typical 1D and 2D 31P, 1H NMR spectra from a representative boreal humus soil are shown in Figure 2. Both 1D and 2D spectra require approximately the same amount of experiment time. The direct comparison between 1D 31P NMR spectra from the sample with (thin line) and without (bold line) sulfide pretreatment emphasizes the marked improvement in the NMR line widths and spectral resolution due to the sulfide.

1D and 2D 31P, 1H NMR spectra of an extract originating from a humus soil sample in a boreal forest approx. 25 km N of Umeå, Sweden. The displayed spectral region contains signals from orthophosphate monoesters. The bold 1D 31P NMR spectrum originates from a soil extract without sulfide treatment, while the thin 1D and the 2D 31P, 1H NMR spectrum were obtained from a sulfide-treated extract. The chemical structures of scyllo-inositol hexakisphosphate (A) and of the lipid hydrolysis product α-glycerophosphate (B) are shown as inserts; the bold letters indicate the P–O–CH2 moiety that gives rise to the marked cross peak based on an interaction between the P and H atoms (Vestergren et al., 2013, unpublished).
The advantage of 2D over 1D NMR becomes most apparent in regions with spectral overlaps, where attributing peaks in 1D 31P spectra to single P compounds can be unreliable. In contrast, overlap is unlikely in the corresponding 2D spectra. For instance, in the 2D NMR spectrum the 31P overlap of scyllo-inositol hexakisphosphate (marked A) and β-glycerophosphate (marked B) with two other P species (labelled 1 and 2, respectively) has been removed by the well-resolved difference in their 1H chemical shifts (Fig. 2). Furthermore, the molecular structure of α-glycerophosphate, where the P–O–CH2 moiety gives rise to a cross peak in the 2D spectrum due to the interaction (so-called “J coupling”) between the P and H atoms, is shown. The resulting compound-specific fine structure in the 1H dimension can also be used for compound identification. Even if cross peaks overlap in both dimensions, this fine structure would still allow detection of two compounds. In 1D 31P NMR, identification of P species is further compromised by the dependence of 31P chemical shifts on pH and the sample matrix. Nonetheless, this problem is also solved using 2D spectra, because the combination of 31P and 1H chemical shifts and the 1H signal fine structure make identification much more reliable. Recently, Vincent et al. (2013) successfully used 2D NMR spectroscopy to characterize soil Po transformations in humus horizons across a 7,800 y-old chronosequence in N Sweden. They reported that the abundances of α- and β-glycerophosphate, nucleotides, and pyrophosphate were higher at the youngest site, whereas DNA, 2-aminoethyl phosphonic acid, and polyphosphate increased up to 1,200 to 2,700 y and then declined thereafter.
In summary, 31P NMR is still the method of choice for soil Po research. To fully explore the advantages of 2D 31P, 1H NMR spectroscopy for improved P species quantification, it is necessary to develop P compound-specific sensitivity factors for 2D NMR and apply advanced chemometric methods to manage the huge datasets (McKenzie et al., 2011).
3.2 Vibrational spectroscopic techniques
Infrared (IR) spectrometry relies on the absorption of IR light with different energy or wavelength (λ), by sample compounds. The wavelength can also be expressed as wavenumber v (with ν = λ−1); i.e., the number of waves in a wave train of 1 cm length (unit of measure: cm−1). If the vibrational or rotational energy of a molecule is enhanced upon absorption of IR radiation, it is described as IR active. This applies to a vast number of molecules, except highly symmetrical ones with symmetrical stretching; these, however, are Raman active. Raman spectroscopy is based on the scattering of light (see below). IR spectroscopy detects molecule vibrations if the electric dipole moment changes during the transition, whereas with Raman spectroscopy vibrations are detected if the polarizability of the molecule is changed during the vibration. In highly symmetrical molecules the vibrations are either IR or Raman active. Whereas IR relies on light absorption, for Raman spectroscopy a monochromatic laser beam of frequency υ0 induces molecular transitions to virtual excited states. The excitation frequency (υex) is emitted when the molecule relaxes to the ground state. In addition to this elastically scattered radiation (Rayleigh scattering), in-elastically scattered radiation is also detected (Raman bands: υ) resulting from the vibration (and rotation) of the scattering molecules. The difference between the frequency or the wave number of the Raman bands and the excitation frequency gives the Raman spectrum (Stokes scattering: υex – υ; and anti-Stokes scattering: υex + υ). In normal Raman spectroscopy only the Stokes scattering is used. Based on the energy or wavelength used to enhance the molecular vibration or rotation, IR analyses can be distinguished into visible (VIS; 400 to 800 nm or 25,000 to 12,500 cm−1), near-infrared (NIR; 800 to 2,500 nm or 12,500 to 4,000 cm−1) or mid-infrared light ranges (MIR; 2,500 to 25,000 nm or 4,000 to 400 cm−1). The spectra are dominated by electronic transitions (VIS), stretching and bonding of NH, OH, and CH groups (MIR), and overtones and combination modes of two or more fundamental vibrations, which may even occur in the MIR range. This range tends to be frequently superior to NIRS in spectral assignment to soil properties (Viscarra Rossel et al., 2006). IR spectroscopy has been used to predict various soil properties such as soil water content, texture, CEC, or soil total C, N contents, as reviewed, e.g., by Soriano-Disla et al. (2014).
The classical method for measuring IR spectra of solid material is the transmission technique where the samples are shone through as thin pellets. For pressing these pellets, often K-bromide has to be added. As well as transmission, reflectance techniques also can be used for infrared spectroscopy. If the preparation of the thin pellets is impossible or too time consuming, the “Diffuse Reflectance Infrared Fourier Transform” (DRIFT) technique can be used. The ground sample is placed in a special holder and the diffuse reflected light is measured. This technique can also be combined with a sample plate to enable high-throughput screening. Solid and liquid samples can be measured using Attenuated Total Reflectance IR spectroscopy (ATR-IR); for this, the IR-beam is introduced into a crystal at an angle exceeding the critical angle for internal reflection and thus reflects at least once at the internal surface in contact with the sample. Depending on the spectral device, dispersive and Fourier-Transform-(FT) spectrometry can be distinguished. Dispersive spectrometry uses prisms or grates to separate the light into the different wave lengths or wavenumbers; whereas FT spectrometry measures all wave lengths simultaneously as an interferogram which is transferred into the spectrum by a Fourier transformation. This technique is currently considered the state-of-the-art especially if synchrotron-based sources are used to aquire the spectra (e.g., Solomon et al., 2005).
3.2.1 Near-Infrared-Spectroscopy (NIRS)
Near-Infrared-Spectroscopy (NIRS) is a standard quality assurance method in many industries (Burns and Ciurczak, 2001; Roberts et al., 2004). Over the past decade, a considerable amount of research has been done to develop NIRS applications for soil analysis (for a general overview see review by Nduwamungu et al., 2009) and models to predict, for example, biological and chemical soil parameters (Chang et al., 2001; Ludwig et al., 2002; Zornoza et al., 2009) and the composition and origin of litter and roots (Lei and Bauhus, 2010; Gruselle and Bauhus, 2010). Phosphorus or phosphate cannot be detected directly with NIRS because of the low dipole moment between P and O. However, P can be quantified via NIRS if it is bound organically or is tightly associated with other soil properties. Therefore, it is not surprising that only few NIRS models are available to predict concentrations of Pt or different P fractions in soil (Chang et al., 2001; Dunn et al., 2002; Malley et al., 2004; Abdi et al., 2012).
Generally, the quality of NIRS prediction models is assessed using a number of statistical parameters comprising the goodness of fit, r2 (measured vs. predicted), root mean standard error of cross validation (RMSECV) and ratio of standard deviation to RMSECV (RPD, Ratio of Performance to Deviation). The prediction quality of models increases with r2 and RPD. Conventionally, it is claimed that models with r2 > 0.81 and RPD > 3 provide acceptable predictions of P; models with r2 < 0.5 and RPD < 2 provide only poor predictions (Chang et al., 2001; Malley et al., 2004; Nduwamungu et al., 2009). For example, NIRS prediction models for Mehlich-III extractable P reached only an r2 of 0.4 and an RPD of 1.18 (Chang et al., 2001). According to a review by Malley et al. (2004), the quality of NIRS-prediction models for extractable P and Pt ranged from very poor (r2 = 0.18, RPD = 1.1 for bicarbonate P) to fair (r2 = 0.81, RPD 2.3 for total Pt). Nonetheless, ongoing work at the University of Freiburg, Germany, is exploring the potential of NIRS to investigate soil P pools of different availability; the procedures and preliminary findings are described here. Soil samples were dried to reduce the impact of the strong influence of the O–H bond on near IR absorption (Malley et al., 2004), and subsequently ground. Spectra were recorded using a Tensor 37 spectrometer from Bruker Optics (Ettlingen, Germany) at constant temperature. This instrument allows continuous measurements of the total NIR-spectral range from 12000 to 4000 cm−1 within one scan, and uses diffuse reflection with an integrating sphere detector. The whole NIR spectral area was scanned with intervals of 16 wave numbers (each interval 64 times) and converted via a Fourier-Transformation into one spectrum. A mean spectrum was created for each sample from 5 replicate measurements. Reference data for soil P in fractions of different availability were determined using the sequential extraction method according to Hedley, modified by Tiessen and Moir (2007). For model development, reference values and spectra were evaluated by means of a Partial Least Square Regression (PLSr) analysis (Conzen, 2005). This chemometric method takes advantage of correlative relationships between the spectral signatures and soil attributes by selecting successive orthogonal factors, which maximize the covariance between the predictor spectra and the laboratory data. Usually, the first derivative of a spectrum is taken to split it into latent variables using PLSr analyses, which are then included in a regression model for estimating the soil properties. For more details on the PLSr models and their use in soil science, the reader is referred to Viscarra Rossel et al. (2006) and citations therein. It should be noted, however, that each PLSr model requires a calibration data set, which may be soil-type specific. Recently, Siebers et al. (2013) have also used artificially produced mathematical data sets to calibrate a PLSr model for XANES spectra evaluation (cf. chapter 3.6), but to the best of our knowledge, similar approaches have not yet been used for PLSr applications to VIS-NIR-MIR analyses in soil science.
Vis à vis the aforementioned work at the University of Freiburg, Germany, the process of NIRS-model development included the following steps: determination of reference values by standard P extraction procedures and wet-chemical methods, measurement of NIR spectra (Fig. 3a), data preparation, selection of the calibration data set, and development of statistical prediction models. The data calibration and model development steps utilize, for example, multivariate calibrations, selection of the best fitting model (Fig. 3b), and evaluation of the model with an independent data set (Chang et al., 2001; Conzen, 2005). As a rule of thumb, at least 150 reference samples should be available for complex samples like soil. Additionally, at least 30 to 40 samples, which are not part of the calibration process, are recommended for model validation.

(a) Examples of typical NIR absorption spectra from soil samples (Cambisols). (b) Model calibration results, predicted vs. measured, for NaOH soluble organic P (Po) (Niederberger et al., 2013, unpublished).
Preliminary NIRS models (Fig. 3b) for Po fractions had acceptable quality (r2 = 0.85 and RPD = 2.61) but not for Pi fractions. Provided that a sufficient number of reference data is available, the assessment of a variety of parameters is possible with one spectrum. However, the more complex the sample is, the more interference and overlapping of spectral information can occur. Therefore, developing NIRS-models for estimation of P fractions in soil that contain a wide variety of other organic compounds is challenging. Although some reference analysis is necessary for the calibration, the main advantage of NIRS is the possible replacement of extractions and wet-chemical fractionations by a faster and operationally simpler method. This makes it especially interesting for large scale surveys with many samples, e.g., in monitoring programs.
3.2.2 Mid-infrared spectroscopy (MIRS)
Starting in the late 1990's, MIRS has been used to estimate the contents of plant-available P (Janik et al., 1998; Daniel et al., 2003; Viscarra Rossel et al., 2006) and Pt (Minasny et al., 2009; Shao and He, 2011) in soil. Viscarra Rossel et al. (2006) noted that MIRS was superior to NIRS, particularly for available P analyses; yet, the calibration functions for both IR methods still lacked high r2 values. Subsequently, Moron and Cozzolino (2007) reported that higher r2 values (at least up to 0.61) could be obtained for estimates of plant available and extractable P if using suitable reference sampling sets. Coefficients of determination exceeding 0.90, however, could only be reached by spiking phosphates to sand fractions of different size (Bogrekci and Lee, 2005a). Rückamp et al. (2012) succeeded in applying MIRS to the rapid assessment of different extractable P forms (i.e., NaHCO3-extractable P for labile P and concentrated HCl-extractable P for stable P) in tropical soils. Indeed, the authors found reasonable coefficients for the prediction of the NaHCO3-extractable Pi and Po (= labile or plant available P bound to mineral surfaces; r2 = 0.70 to 0.74), and the HCl-extracted P (= stable Pi associated with Ca or occluded in sesquioxides; r2 = 0.86). However, it was not possible within this sequential fractionation to estimate successfully the contents of stable Po in the same concentrated HCl extract (Negassa and Leinweber, 2009) by MIRS and PLSr. Nevertheless, further progress in this research is expected due to advances in spectrometer hard- and software, and more effective mathematical spectra analysis that is based on support vector machines, wavelets and/or neural networks (e.g., Daniel et al., 2003; Odlare et al., 2005). For example, Daniel et al. (2003) used neural networks to estimate P from the VIS-NIR spectrum. Since then, however, the use of PLSr has become prevalent.
Strong P–O–(H) stretching and bending vibrations are generally observed in the 1250 to 750 cm−1 spectral range; this has allowed the use of IR to identify reaction products of P fertilizer in soil (Beaton et al., 1963) and different phosphate minerals, e.g., hydroxyapatite, and octacalcium phosphate (reviewed by Kizewski et al., 2011), and to reveal through sorption studies onto Al and Fe oxides, for example, the nature of P bonds in soil (cf. chapter 4.2; Arai and Sparks, 2001; Luengo et al., 2006, Elzinga and Kretzschmar, 2013; Waiman et al., 2013). Moreover, certain absorption bands have been shown to be characteristic for certain P compounds; for example, the 1,090 and 1,150 cm−1 spectral range for Al- and Fe-phosphate, respectively, or at 1,100 cm−1 for humic P (He et al., 2006a; 2006b). Phytic acid exhibits pronounced signals at 968, 1,008, and 1,052 cm−1; this triplet shifts to higher wave numbers in the presence of metal phytates. The P–O–C stretch vibrations in phytate resulted in a hybrid spectral feature and peaking triplet signals from 790 to 900 cm−1; to date, this signature is unique and, as such, has been proposed to distinguish phytates from other P compounds (He et al., 2007). Remarkably, with suitable sample pretreatment (e.g., extraction of biological soil materials) it has been possible to detect polyphosphate P as a broad signal in the 1,200 to 1,400 cm−1 regionusing ATR-FTIR spectroscopy in combination with PLSr analyses (Koshmanesh et al., 2012).
In summary, IR spectroscopy (NIRS and MIRS) has been reasonably successful in identifying different P species in environmental samples and, in combination with PLSr or related mathematical techniques; it has gained increasing potential for minimum-invasive, high throughput soil P sensing. Nonetheless, it is still unclear whether successful IR-based evaluations of P bonding forms are always the result of specific vibrations in the MIR or NIR spectra, or the consequence of poorly understood or even spurious relationships with other soil parameters, such as Fe-oxides and soil OM quality.
3.2.3 Raman spectroscopy
In P research, Raman spectroscopy has been used mainly to understand the nature and importance of chemical bonds, both in P-bearing minerals and between P species and reactive surfaces. The main P containing substances analyzed by Raman spectroscopy are hydroxyapatite [Ca10(PO4)6(OH)2], octacalcium phosphate [Ca8H2(PO4)6 · 5 H2O] minerals and phosphate adsorbed on Fe- and Al-oxides. Most Raman measurements are usually carried out with pure minerals or model systems, with only a few using actual soil samples because of the interference by fluorescence. The intensity of the Raman signals resulting from the inelastic scattering depends on the changing of the molecular polarizability during the vibration; this goes with the second power and, fortunately, the P–O symmetric stretching of phosphate is very well detectable using Raman spectroscopy. The wave number is in the region of about 950 cm−1, depending on the surroundings of the PO4 triangular pyramid (Lanfranco et al., 2003; Alvarez et al., 2004).
In general, Raman spectroscopy is an analytical method that can be employed in the laboratory using probes both for solid and liquid states, whilst portable Raman sensors for use in the field are also vailable described to measure P in soil (Bogrekci and Lee, 2005b; Lee and Bogrekci, 2006). The main advantages of Raman measurements include the simple sample preparation, the small amount of analytical material required and the fact that the resulting data can be used without any reprocessing. If the matrix is very heterogeneous and fluorescent substances are present, extraction steps can be undertaken to isolate the analyte. In pure systems, various band frequencies of different endmember elements (Ca, Cd, Pb–Sr) of hydroxyapatite can be identified. In soils, Raman spectroscopy was able to trace added hydroxyapatites at a sub-micron grain size and at concentrations down to 0.1% (w/w); this is below the routine detection limits of X-ray diffraction analysis (Lanfranco et al., 2003). Another advantage is the possibility to analyze aqueous solutions—owing to the very weak Raman scattering of water. Due to the excitation using a laser beam, only a very small area of the probe is analyzed. Therefore, Raman spectroscopy can be combined with microscopic techniques (“µ-Raman”). Combinations with optical microscopes (normal and confocal Raman microscopy) are currently available, as well as those with non-optical microscopic techniques which have a higher resolution and are not limited by the wave length of visible light (e.g., AFM: Atomic Force Microscopy; SNOM: Scanning Near-Field Optical Microscopy). Cusco et al. (1998) used µ-Raman to differentiate hydroxyapatite and β-tricalcium phosphate by means of their OH− groups as well as their bands from internal PO modes. Lanfranco et al. (2003) used µ - Raman in combination with a microscope to identify mixed - metal phosphates in phosphate - remediated metal - contaminated land. Kasioptas et al. (2011) followed the 18O isotope incorporation in apatite using a combination of Raman spectroscopy and microscopy. A relatively new application is the use of IR and Raman spectroscopy for mapping and imaging (Salzer and Siesler, 2009). Recently, phosphate phases in sewage sludge ash - based fertilizers were investigated by a confocal µ - Raman spectrometer (Vogel et al., 2013a) and a microscope coupled with a synchrotron - based radiation IR spectrometer (Vogel et al., 2013b).
modes. Lanfranco et al. (2003) used µ - Raman in combination with a microscope to identify mixed - metal phosphates in phosphate - remediated metal - contaminated land. Kasioptas et al. (2011) followed the 18O isotope incorporation in apatite using a combination of Raman spectroscopy and microscopy. A relatively new application is the use of IR and Raman spectroscopy for mapping and imaging (Salzer and Siesler, 2009). Recently, phosphate phases in sewage sludge ash - based fertilizers were investigated by a confocal µ - Raman spectrometer (Vogel et al., 2013a) and a microscope coupled with a synchrotron - based radiation IR spectrometer (Vogel et al., 2013b).
To date, Raman spectroscopy has not been used extensively to characterize soil-inherent P because of the interference of fluorescence effects. The mechanism of fluorescence is very similar to the underlying mechanism of the Raman scattering, but it is much stronger (Kizewski et al., 2011). Fluorescing substances therefore disturb the Raman spectra, and among these distorting compounds are all naturally organic materials, thus limiting its application for soil samples. Excitation in the IR frequency range with a FT-spectrometer can remedy this interference because the power of Raman emission increases with the fourth power of the frequency of the source (FT-Raman Spectroscopy) (Aminzadeh, 1997). Also, surface-enhanced Raman spectroscopy (SERS) has been used to minimize these problems (Francioso et al., 1996; 1998), as has shifting the excitation energy to high energy ranges (i.e., ultraviolet (DUV Raman spectroscopy)—first examined by Asher and Johnson (1984).
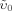 of (P16O4)3− the wave numbers of the new bands
of (P16O4)3− the wave numbers of the new bands 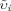 (i = 1, 2, 3, 4) can be calculated for phosphate molecules if one to four O atoms are replaced by 18O isotopes (Smith and Carabatos - Nédelec, 2001; Kasioptas et al., 2011):
(i = 1, 2, 3, 4) can be calculated for phosphate molecules if one to four O atoms are replaced by 18O isotopes (Smith and Carabatos - Nédelec, 2001; Kasioptas et al., 2011):
 ()
()m16 and m18 are the masses of the two O isotopes.
To test the potential of this approach, a heating experiment with 18O enriched silver phosphate (Ag3P18O4) was conducted. Silver phosphate was used because this compound is commonly used to extract phosphate from soil matrices in O-isotope studies (cf. chapter 4.6). The 18O-phosphate was heated for 16 h at 600°C in normal atmosphere to forcibly test the rate of exchange of 18O by 16O at enhanced temperatures; the Raman spectroscopic measurement was performed with a BRUKER Fourier Transform (FT)-Raman spectrometer “RFS 100 S”. The excitation occurred in the IR range (1,064 nm) using a Nd:YAG-Laser (power: 200 mW). Fig. 4 shows the spectrum of the heated Ag3P18O4. The signals at 910 cm−1 and 859 cm−1 agree very well with the measured wave numbers of the symmetric P–O stretching mode of pure Ag3P16O4 and Ag3P18O4, respectively. The observed wave numbers of the bands are in a good agreement with the calculated values with Eq. 1.

Raman spectrum of heated Ag3P18O4 and the respective peak shifts due to successive 18O exchange (Lewandowski and Amelung, 2013, unpublished). The color image of this figure is in the digital version of this article.
This example shows that Raman spectroscopy is a powerful tool for detecting and tracking the pattern of position-specific 16/18O exchange reactions within phosphate-bearing materials. While the shift of the signal's wavenumber is directly correlated to the number of the exchanged atoms (see Eq. 1) the peak area is proportional to the ratio of this group in the probe. Therefore, Fig. 4 demonstrates that a significant proportion of the 18O isotopes are already exchanged by 16O, but that some of the phosphate groups still contain of 18O isotopes. In molecules with phosphate groups, the O atoms are not equal and the exact position of the Raman signals also depends on the position of the isotope exchange. This can be shown by quantum-chemical calculations of the peak positions of the O isotopes at different positions in the phosphate. In real Raman spectra these differences are difficult to detect because the signals are broadened by matrix effects. Raman spectroscopy has also been used to observe mineral replacement reactions, such as the transition of aragonite and calcite into hydroxyapatite by hydrothermal treatment with (NH4)2HPO4 (Kasioptas et al., 2011) and the replacement of aragonite by calcite (Perdikouri et al., 2011). In both cases the incorporation of the heavier O isotope into the apatite and carbonate, respectively, was monitored. Furthermore, the exchange kinetics of O isotopes between water and CO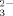 was studied (Geisler et al., 2012). In all measurements the Raman band of the symmetric vibration of the phosphate or carbonate molecule was the tracer of the isotope exchange mechanism.
was studied (Geisler et al., 2012). In all measurements the Raman band of the symmetric vibration of the phosphate or carbonate molecule was the tracer of the isotope exchange mechanism.
In summary, Raman spectroscopy is an excellent and novel but as yet underexplored tool in the determination of soil P composition and dynamics. Although the main applications make effective use of model systems, new exciting possibilities in spatial imaging lend the technique to more heterogeneous systems (Lanfranco et al., 2003).
3.3 High resolution mass spectrometry
Mass spectrometry (MS) uses the mass-to-charge ratio (m/z) of charged molecules or molecular fragments to obtain a ‘‘chemical fingerprint'' of a sample and to characterize its molecular-chemical composition. For soil samples some extraction is usually required as a precondition for MS analysis. Extracts are either separated by chromatography or directly injected into the mass spectrometer. Various ionization techniques exist, such as chemical ionization at atmospheric pressure (APCI), matrix assisted laser desorption ionization (MALDI or Laser ablation, LA-ICP-MS; cf. chapter 4.4) and electro spray ionization (ESI); among these, the latter has been most frequently used in Po research. ESI is a soft ionization technique because of minimal fragmentation which facilitates the characterization of intact molecules (Banerjee and Mazumdar, 2012). It is very sensitive for polar compounds; however, in the positive mode it often leads to multiple charged ions and adducts with alkaline metals, making the determination of molecular species and, thus, spectral evaluation, difficult. For P analysis this is even more challenging because of the poor ionization efficiency of P-containing compounds resulting in low sensitivity (Reemtsma, 2009). Very low concentrations of P-containing molecules, e.g., 0.2 to 0.3% P in humic acid (He et al., 2006b) against a dominant background of dissolved organic C-, N- and O-compounds add to the analytical problems. Therefore, selective isolation and pre-concentration steps to increase P concentration and remove interfering non-P containing dissolved organic matter (DOM) molecules is usually required. Examples are extraction and concentration by solid phase extraction and/or dialysis (Tfaily et al., 2012), tangential cross flow filtration or P precipitation (Llewelyn et al., 2002; Cooper et al., 2005), liquid chromatography (e.g., De Brabandere et al., 2008) and high-performance size exclusion chromatography (El-Rifai et al., 2008).
High resolution mass spectrometry can distinguish among different mass-to-charge ratios within very complex mixtures that comprise upwards of several thousand compounds. Due to the high mass accuracy of usually < 1 ppm, this technique is able to assign sum formulas to certain signals (Cooper et al., 2005; Reemtsma, 2009). However, the majority of significant high resolution MS studies do not include P in their sum formula calculations and m/z signal assignments, since this greatly increases the number of chemically possible sum formulae (i.e., 7,087,578 considering C, H, O, N, and S vs. 32,238,738 considering C, H, O, N, S, and P in the range from 1 to 1,000 Da). Furthermore, a verification of the assigned sum formula by stable isotopic patterns, as often performed for C, N, and S containing compounds, is impossible because 31P is the only stable P isotope (Koch et al., 2007). Therefore, spectra evaluation has not yet routinely included P, although its inclusion does provide a more complete chemical characterization of the sample. For instance Ohno and Ohno (2013) showed that the number of assigned m/z peaks could be increased by approx. 11% after including P in sum formulae calculation. In order to resolve the very small difference in m/z signals required for such signal assignments, very high resolution (R > 61,000) mass spectra are required; e.g., the exchange of a P–H with an O2-group results in a difference at one nominal mass of only 8.24 mDa. Only a few devices can provide this high mass resolution for large molecules such as organic phosphates, i.e., Orbitrap-, multi-reflectron Time-of-Flight (TOF) or fourier transform ion cyclotron resonance (FT ICR) mass spectrometers. FT ICR-MS devices deliver unbeaten resolving power and semi-quantitative determination by using a very strong magnetic field to trap the ions inside a vacuum cell (Marshall et al., 1998). Orbitrap-MS devises use very strong electric fields and spindle shaped electrodes and TOF-MS use long flight paths to enable quantitative high resolution measurements with high scan rates (Hu et al., 2005; Ballesteros-Gomes, 2013).
Ultrahigh resolution and mass accuracy provided by such MS devices enables the simultaneous detection of thousands of individual m/z signals in a sample, each representing a unique molecular formula. Therefore, an extensive treatment of spectral data is needed to reduce complexity, and to examine, interpret and visualize this massive molecular-chemical data set with the aim to identify spectral differences among samples. Most often, van Krevelen diagrams are used for presentation of high resolution mass data as they enable visual differentiation of compound classes based on their O/C and H/C ratios of molecular formulae (e.g., Wu et al., 2004; Abdulla et al., 2013) (Fig. 5).

Van Krevelen diagram of assigned molecular masses (m/z) of dissolved organic matter (DOM) samples measured by FT ICR-mass spectrometry. Modified replot from Abdulla et al. (2013) with permission of ACS Publications. C, H, N, O, S, and P were considered in the sum formula calculations assignment. As an example the insert (lower right) shows all assigned masses with the same nominal mass m/z 369 which could be resolved only by high resolution mass spectrometry. A color image of this figure is in the digital version of this article.
Recently, Abdulla et al. (2013) used two-dimensional (2D) correlation of van Krevelen diagrams, m/z signal intensities, as well as 2D hetero-correlations between m/z signal intensity changes and peaks in the corresponding NMR spectra, to reveal differences and similarities in DOM samples along a salinity transect through the Elizabeth River/Chesapeake Bay system to the coastal Atlantic Ocean of Virginia, USA. Multivariate statistical analysis methods such as principal component analysis or hierarchical clustering analysis also can be used for spectra evaluation (Hur et al., 2010). The application of high resolution MS in soil science has facilitated the non-targeted characterization of C, N, O, and H compounds in DOM (e.g., Kujawinski et al., 2002; Reemtsma, 2009). However, only a few studies have looked at the composition of P containing compounds in, e.g., surface water (Llewelyn, 2002; Abdulla et al., 2013) or DOM from anthropogenic sources (i.e., runoff, urban soil) (Killberg-Thoreson et al., 2013). Conversely, targeted mass spectrometry at normal or high resolution (roughly 10 to 50 times lower than ultrahigh resolution provided by the FT ICR-MS devices, described above) are commonly used to quantify natural and anthropogenic P-containing compounds in soils, e.g., inositol phosphates (reviewed by Cooper et al., 2007), phospholipids (cf. chapter 3.4), flame retardants (Ballesteros-Gomes et al., 2013) and, particularly, pesticides (Börjesson et al., 2000; Ibáñez et al., 2005; Hanke et al., 2008).
Among others, glyphosate [N-(phosphonomethyl)-glycine] is becoming one of the most intensively studied herbicides; this is because non-selective, glyphosate-based herbicides such as Roundup®, Nufarm® and Touchdown® are among the most widely used herbicides around the world (Steinmann et al., 2012). Several studies have used MS based techniques to investigate glyphosate and aminomethylphosphonic acid (AMPA, glyphosate's primary metabolite) concentrations in soils (e.g., Todorovic et al., 2013; Aparicio et al., 2013). For example, Aparicio et al. (2013) reported that glyphosate and AMPA concentrations in various Argentinian agricultural soils of different types and land use systems ranged between 35 and 1502 μg kg−1, and 299 to 2256 μg kg−1, respectively. General overviews on the mobility, leaching and fate of glyphosate have been published by Vereecken (2005) and Borggaard and Gimsing (2008).
The diverse functional groups of glyphosate and its primary metabolite, aminomethylphosphonic acid (AMPA), result in molecules with amphoteric characteristics and high polarity; these structural properties are essential for the analysis of such substances (Martínez Vidal et al., 2009; Stalikas and Konidari, 2009). Recent analytical methodologies for glyphosate and AMPA include, besides others, GC-MS (Börjesson et al., 2000) and liquid chromatography (LC) coupled to tandem mass spectrometry (LC-MS/MS) (Ibáñez et al., 2005; Hanke et al., 2008; Botero-Coy et al., 2013). Because of low molecular masses and ionic characteristics together with high solubility in water, a pre-column derivatization with 9-fluorenylmethoxycarbonylchloride (FMOC-Cl) is required to enable chromatographic separation (Hanke et al., 2008; Sanchís et al., 2012) (Fig. 6a). By subsequent separation using a reverse phase LC-system, sufficient retention times with good resolution can be achieved (Fig. 6b). Following column elution, the compounds are detected by ESI-MS/MS. The use of the “Selected Reaction Monitoring” mode enhances the detector sensitivity as the number of parent and product ions is limited (Fig. 6c). Together with stable isotopes of glyphosate and AMPA, that serve as internal standards, this approach may yield quantitatively reproducible data with high precision and accuracy in quantification (e.g., LC-GC MS/MS: 50 ng L−1 for water and 0.05 mg kg−1 for soil; Ibáñez et al., 2005).

(a) Precolumn derivatization of glyphosate and AMPA: the reaction of FMOC-Cl with glyphosate and AMPA results in the formation of glyphosate-FMOC and AMPA-FMOC with molecular masses of 391 and 333 g mol−1, respectively. (b) Total Ion Chromatogram of the FMOC-derivatives: the derivatized compounds are separated on a reverse phase liquid chromatography system and detected through Tandem Mass Spectrometry after Electrospray Ionization. (c) Operation in the Selected Reaction Monitoring mode allows detection of the compounds based on the m/z ratios of their product ions. The isotope-labelled glyphosate–1,2–13C2–15N and AMPA–13C–15N serve as internal standards (Abraham, 2014, unpublished).
Inductively coupled plasma mass spectrometry (ICP-MS) is another methodological approach to characterize and quantify P in environmental samples. In contrast to the aforementioned soft ionization techniques, the very high energy impact of the plasma fragments molecules to their elemental composition, thereby enabling P determination by monitoring the m/z signal of 31P+ (m/z: 30.974). Although ICP-MS has been used mainly for total element quantification in liquid samples, instrument developments and combinations with various separation and sample introduction techniques (e.g., HPLC, GC, laser ablation) have continuously improved its capabilities (Pröfrock and Prange; 2012). For example, the development of collision (CC-ICP-MS) or dynamic reaction (DRC-ICP-MS) cells as well as the availability of high resolution ICP-MS have reduced interference from isobaric polyatomic ions (e.g., 15N16O+: m/z 30.9950; 14N16O1H+: m/z 31.0581) and, thus, improved sensitivity. Furthermore, laser ablation (LA-ICP-MS) has enabled spatially resolved P analysis in solid samples (Santner et al., 2012; cf. chapter 4.4), and coupling the ICP-MS with a preceding chromatographic separation technique allows the characterization and quantification of Po compounds (e.g., Vonderheide et al., 2003; Richardson et al., 2006; Chen et al., 2009).
The selected examples of mass spectrometry presented here demonstrate the vast potential of the technique in investigating P compounds in soil, especially in the soil solution. As an untargeted MS method, ultrahigh resolution FT ICR-MS is still in its early stages as a tool for analysing soil P. With an increasing number of instruments becoming available in recent years and in the near future, more applications can be expected. For targeted MS methods, the ever increasing sensitivity of instrumentation enables deeper insight in the fate of specific P compounds in the soil–plant and soil–water systems.
3.4 Direct Infusion Nanospray Quadrupole Time-of-Flight Mass Spectrometry (Q- TOF MS/MS) to quantify intact phospholipids
Around 15 to 80% of the total P in soils occurs in organic form (Stevenson, 1994); thereof 3 to 24% is bound in the microbial biomass (Brookes et al., 1982). However, soil microorganisms constitute not only a substantial part of P in the soil, but are also involved in a number of important processes in the soil P cycle, e.g., mineralization and immobilization of P (e.g., Oberson and Joner, 2005; cf. chapter 4.3). The dominant forms of P in microorganisms are nucleic acids and phospholipids, which account together for approximately 60% of total Pmic (Bünemann et al., 2011). Phospholipids are essential membrane components of all living cells and are characterized by a rapid degradation in soils after cell death due to enzymatic hydrolysis. Hence, the remaining amount of intact phospholipids in soil is highly correlated with the living soil microbial biomass and can serve as a quantitative measure of the Pmic (White et al., 1993). Although phospholipids represent only a small proportion of the total Po in soils (≈ 0.5 to 7%; Stevenson, 1994), they are important elements of the soil P cycle and fertility, since their rapid synthesis and degradation cycles allow a significant quantity of mineralized P to become and stay plant available.
Phosphatidic acid (PA) is one of the simplest glycerophospholipids, consisting of two fatty acids esterified to the sn-1 and sn-2 position of a glycerol backbone and a polar phosphate headgroup which is attached to the sn-3 position. The diacylglycerol moiety of phosphatidic acid is incorporated into different phospholipids by phosphate ester condensation with different alcohols [e.g, phosphatidylserine (PS), phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylglycerol (PG) and phosphatidylinositol (PI)]. In addition to the ester-linked glycerophospholipids there are also soil bacteria that occur ubiquitously in soil having ether-linked glycerophospholipids as membrane lipids which are characterized by an ether-linked acyl chain at the sn-1 or sn-2 position. These ether bonds are more resistant to oxidation and high temperatures than ester bonds, and ether bound phospholipids are most prominent in Archae (Albers et al., 2000). The distribution of intact polar branched tetraether lipids in peat and soil have been studied (e.g., Peterse et al., 2011), and their soil environmental importance reviewed by Schouten et al. (2013).
The co-existence in soil of such a broad range of phospholipid structures is explained by the fact that the different headgroups can be combined with a large number of fatty acids that vary both in chain length and degree of desaturation. Since the fatty acid composition in phospholipids varies widely among different microorganisms, their distribution profiles reflect the soil microbial community structure (White et al., 1993). Therefore, phospholipid fatty acid (PLFA) profiles are widely used as biomarkers of the microbiota in soil (Buyer and Sasser, 2012). For PLFA analysis by gas chromatography (GC), the extracted and separated phospholipids are methanolyzed and the acyl groups are cleaved and converted into their methyl esters. In this approach only the acyl groups, but not the headgroups, can be analyzed. To obtain further structural information within the different phospholipid classes, separation by thin layer chromatography (TLC) prior to fatty acid methyl ester (FAME) synthesis is required (Wu et al., 1994). Silica gel is most commonly used for this purpose, in combination with different solvent systems (Kahn and Williams, 1977). Once separated, the acyl groups can be quantified by isolating individual lipids from the silica material (Benning and Somerville, 1992). However, GC-based methods are unable to provide information about the different molecular species of phospholipids. As a consequence, further analytical methods have been developed to investigate intact phospholipid molecular structures, as reviewed by Peterson and Cummings (2006).
In addition to chromatography-electrospray ionization-mass spectrometry (LC-ESI-MS; Zhu et al., 2013), quadrupole time-of-flight mass spectrometry (Q-TOF MS/MS) represents a new strategy to characterize the phospholipids molecular species in their native structure. As a solution state method, the Q-TOF MS/MS measurements require the extraction of phospholipids from the sample.
Total lipids of soil samples (fine earth < 2 mm, ≈ 1 to 5 g wet weight) are extracted in two steps, as described by Bligh and Dyer (1959) with slight modifications. Briefly, formic acid is added to the solvent of the first extraction to prevent lipid degradation by lipases during lipid isolation (Browse et al., 1986). Phase separation after extraction is achieved by adding 1 M KCl / 0.2 M H3PO4. The extracted lipids are subsequently fractionated based on their polarity by solid phase extraction, resulting in an extract highly enriched in phospholipids, which is further characterized by a reduced ion suppression during Q-TOF MS analysis. Afterwards, the polar methanol extract is evaporated under N2 and the remaining lipids are re-dissolved in Q-TOF solvent [chloroform / methanol / 300 mM NH4-acetate (300:665:35); Welti et al., 2002. For quantitative analysis a phospholipid standard mix is used.
These extracts are directly infused via a nano-capillary supplied infusion chip (HPLC chip), ionized by nanospray ionization and separated according to their m/z value. Afterwards, specific ions are selected and characteristic fragment ions of the molecular ions are generated by collision-induced dissociation in the MS/MS mode. In the TOF analyzer, ions are accelerated, separated, and detected at the photomultiplier plate. The quantification of phospholipids is based on precursor ion or neutral loss scanning in relation to internal phospholipids standards of known concentrations (Gasulla et al., 2013). For each phospholipid class at least two different internal standards are selected, which are absent in soil samples, enabling a very precise and reliable phospholipids quantification.
A pioneer application of Q-TOF MS/MS analysis to characterize and quantify the molecular species composition of different phospholipid classes was this review was conducted on soils from four different variants at arable and forest sites (Fig. 7): arable topsoils in crest and depression positions and forest topsoils under pine and spruce. Almost all phospholipid classes (PS, PC, PE, PG, and PI) showed site-specific variations in the total concentrations (Fig. 7a), reflecting differences in their microbial and plant-derived phospholipids. In particular, the molecular species patterns of PC, PE, and PG differ greatly among arable and forest test sites. The concentration of C16:1 and C18:1-containing molecular species of PC and PE are significantly larger in the arable while the concentrations of C18:2-containing molecular species of PC and PE are larger in the forest soils (Fig. 7b). The total concentrations of PC and PE, as most abundant classes of phospholipids in eukaryotic cells, are significantly larger in the slightly acidic to neutral (pH 6.3 to 7.1) fertile arable than in the acidic (pH 4.1 to 4.8) nutrient-deficient forest soils. Oleic acid (C18:1) was mainly present in the di-unsaturated species 36:2 (C18:1/C18:1). It is a main component of PG in diverse plant materials, with increased concentrations in some arable crops (e.g., oilseed rape), but also as a component in conifers (Piispanen and Saranpää, 2002). Furthermore, C18:1 and also C16:1-containing molecular species of PC and PE originate also from bacterial biomass (Frostegård and Bååth, 1996). The C18:2-containing molecular species of PC and PE were described as dominant components of phospholids in litter under pine and spruce (Wilkinson et al., 2002), indicating fungal biomass (White et al., 1996).

Phospholipid composition of soils from four different sites (arable: crest and depression, forest: pine and spruce) in Northern Germany as determined by Q-TOF MS/MS analysis (n = 5). (a) Total concentration of major phospholipid classes (phosphatidylserine, PS; phosphatidylcholine, PC; phosphatidylethanolamine, PE; phosphatidylglycerol, PG; phosphatidylinositol, PI; phosphatidic acid, PA) in nmol mg−1 soil dry weight (DW). (b) Molecular species composition (mol%) of PS, PC, PE, PG, PI and PA. The data present means and standard deviations of five replicas (Siebers et al., 2013, unpublished).
The quantification of phospholipids by Q-TOF MS/MS offers several advantages over the GC-based PLFA analyses: (1) Lower detection limits enable the analysis of small sample amounts. (2) The more straightforward sample purification (avoiding TLC separation and fatty acid methylation) facilitates direct measurements of intact phospholipids from soil. (3) The Q-TOF MS/MS method provides detailed information about the molecular species composition of different phospholipid classes. However, additional GC MS measurements are required to determine the position of the double bond, which is an additional marker in the PLFA analysis (e.g., Klamer and Bååth, 1998). Therefore, it is advisable to combine the novel Q-TOF MS/MS-based approach with complementary GC-based analyses. This would enable a detailed insight into the composition of phospholipids as part of a fast cycling Po pool in soil and helps to disclose changes in the soil microbial community structure and their adaption to altering environmental conditions such as P-deficiencies.
3.5 Nano-scale Secondary Ion Mass Spectrometry (NanoSIMS)
Nano-scale Secondary Ion Mass Spectrometry (NanoSIMS) is a destructive mass spectrometric technique linking high-resolution microscopy with elemental and isotopic analysis. Various papers have emphasized the potential of NanoSIMS in soil science (e.g., Herrmann et al., 2007; Heister et al., 2012; Kilburn et al., 2010; Mueller et al., 2012). However, soil applications of this technique are still in their infancy. The following descriptions refer to the CAMECA NanoSIMS 50L, the latest generation of NanoSIMS spectrometers. During a NanoSIMS analysis the sample surface is bombarded perpendicular with a focused primary ion beam (Cs+ or O−). This leads to the sputtering of the upper surface of the sample and the release of neutral particles and positively or negatively charged mono-atomic or poly-atomic secondary ions (e.g., 13C−, 31P−, 31P16O−, 56Fe16O− when using Cs+ as primary ions). Poly-atomic secondary ions result from recombination reactions of reactive mono-atomic ions (McMahon et al., 2006). The secondary ions are extracted coaxially and accelerated into the double-focusing sector field mass spectrometer, capable of providing the high mass resolution needed, e.g., for the differentiation between 13C14N− (m/z 27.016) and 12C15N− (m/z 27.009). By focusing the primary ion beam, a spatial resolution down to ≈ 150 nm (O−) and ≈ 50 nm (Cs+) (Mueller et al., 2012) can be achieved. However, beam focussing decreases primary beam intensity and, thus, reduces secondary ion yield. Even if the beam is less focused to maintain a sufficient ion yield, a spatial resolution of ≈ 200 nm (Cs+) (Heister et al., 2012) can be achieved, which is above the capability of other lab-based P imaging techniques (e.g., micro-autoradiography: ≈ 0.25 to ≈ 1 mm).
Up to seven m/z signals from secondary ions can be recorded simultaneously using electron multiplier or faraday cup detectors. To prevent atmospheric interference with the primary and secondary ions, the measurements are taken under ultra-high vacuum (down to 1.0 10−11 kPa), prohibiting the analysis of non-vacuum stable samples (e.g., outgassing samples). Regions of interest are analyzed pixel by pixel, yielding pictures of the elemental and/or isotopic composition. Several sequential images from the same spot can be accumulated and merged after drift correction to improve the signal-to-noise ratio.
Static SIMS techniques [e.g., Time of Flight (TOF)-SIMS] use primary ion beams with limited dose (< 1012 ions cm−2) enabling characterization of the molecular composition of the surface monolayer (Boxer et al., 2009). By contrast, in dynamic NanoSIMS that uses a high primary ion beam dose, beam impact results in an almost complete fragmentation of all molecules at the sample surface and, thus, rapid surface erosion. This inherently destructive nature of NanoSIMS enables not only the characterization of the P distribution in the surface monolayer, but also 3-dimensional (3D) mapping by reconstructing the images obtained from the subsequent scanning cycles. However, all analyses are affected by element-, isotope- and matrix-dependent sputtering rates, ion yields, and charging effects (Winterholler et al., 2008; Ghosal et al., 2008) as well as topographical unevenness of the sample. Therefore, absolute quantification as well as the comparison of spatial distribution patterns of different elements by NanoSIMS remain challenges. Attempts to partly overcome such difficulties combine topographical information from atomic force microscopy with NanoSIMS analysis (Wirtz et al., 2013) and the analysis of standard materials of known composition and matrix to determine matrix-specific mass and ion yield fractionation. Due to the very complex nature of soil matrices, ratios between elements/isotopes are preferred or the signal is normalized to a homogenously distributed reference ion (Dérue et al., 2006). More recently, Hatton et al. (2012) used NanoSIMS in conjunction with bulk isotope ratio mass spectrometer (EA/IRMS) measurements on 13C- and 15N-labeled density fractions of soils (internal standard) to calibrate isotope-ratios obtained by NanoSIMS. They demonstrated that this internal calibration yielded more accurate C/N and 15N/14N ratios than the traditional correction method using reference standards. However, this requires that the observation areas in NanoSIMS reflect the bulk macroscopic properties.
Soil samples to be analyzed by NanoSIMS need to be plane (topography of the grains is below ≈ 10 μm) and clean without ablating particles to prevent contamination of the high voltage lens system closely located to the sample. Therefore, soil samples are commonly deposited onto the sample holder by drop coating or embedded in ultra-high vacuum resistant resin, thin sectioned, and subsequently polished to attain the desired plane surface for analysis (Herrmann et al., 2007). However, optimized preparation protocols for soil samples are still under development; essentially, the procedures are not as straightforward as those for biological samples which have been prepared successfully by ultrafast plunge-freezing techniques (e.g., Misevic et al., 2009). To prevent charging during analysis, the prepared samples can be sputtered with gold, platinum, or carbon before loading into the NanoSIMS. In practical measurements, regions of interest are initially selected using the optical cameras, and then these are more accurately observed using the secondary electron image created by scanning the primary ion beam.
NanoSIMS offers the possibility to resolve the 2D- and 3D spatial P distribution in soil with high resolution and precission. This gives the opportunity to localise hotspots of P enrichment or sharp and narrow zones of P depletion around roots or fungal hypae and, thus, helps to spatially resolve P depletion and transfer in the plant-soil system. Although 31P is the only stable P isotope, P can be measured either as 31P− , 31P16O− or 31P16O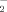 . Since there are no other ions or clusters significantly interfering at these masses, high mass resolution is not required. Thus, high transmission rates can be employed to account for the low ionization efficiency and abundance of P compared to C in most soil samples. The detection limit for P is difficult to determine, since the bulk soil P concentration can be quite misleading with respect to surface P concentrations predominantly accessed by NanoSIMS. However, the detection of most elements in the range of a few mg kg−1 is typically possible (Misevic et al., 2009). Therefore, for soil samples several sequential images are combined to yield images with improved signal - to - noise ratio; an example is provided in Fig. 8b. Fig. 8a, b shows the spatial distribution of 12C14N− (a) and 31P− (b) in free - living N2 - fixing soil bacteria exposed in advance to an 15N2 atmosphere. The same bacteria were also treated with an elevated NaCl (200 mM) solution to simulate salt stress (not shown). Plotting the ratios of 31P/12C and 14N/12C as functions of the 35Cl/12C signal ratio reveal the influence of the salt stress on bacterial N and C uptake (Fig. 8c). The constant values for the ratios 31P/12C and 15N/12C in the bacterial cells in both treatments suggest an adaptation of the bacteria strain to salt stress.
. Since there are no other ions or clusters significantly interfering at these masses, high mass resolution is not required. Thus, high transmission rates can be employed to account for the low ionization efficiency and abundance of P compared to C in most soil samples. The detection limit for P is difficult to determine, since the bulk soil P concentration can be quite misleading with respect to surface P concentrations predominantly accessed by NanoSIMS. However, the detection of most elements in the range of a few mg kg−1 is typically possible (Misevic et al., 2009). Therefore, for soil samples several sequential images are combined to yield images with improved signal - to - noise ratio; an example is provided in Fig. 8b. Fig. 8a, b shows the spatial distribution of 12C14N− (a) and 31P− (b) in free - living N2 - fixing soil bacteria exposed in advance to an 15N2 atmosphere. The same bacteria were also treated with an elevated NaCl (200 mM) solution to simulate salt stress (not shown). Plotting the ratios of 31P/12C and 14N/12C as functions of the 35Cl/12C signal ratio reveal the influence of the salt stress on bacterial N and C uptake (Fig. 8c). The constant values for the ratios 31P/12C and 15N/12C in the bacterial cells in both treatments suggest an adaptation of the bacteria strain to salt stress.

Spatial distribution of (a) 12C14N− and (b) 31P− in free-living N2-fixing soil bacteria, and (c) 31P/12C and 14N/12C ratios as function of the 35Cl/12C ratio of the bacteria without (black) and under salt stress (white) (Vogts and Baum, 2013, unpublished). A color image of this figure is in the digital version of the article.
NanoSIMS does not allow the speciation of individual P compounds. However, spatial correlations with other elements such as Fe, Al or Ca may provide information on the chemical coordination of P. Since 31P is the only stable P isotope, NanoSIMS may have some potential in 32P or 33P tracer experiments to monitor the fate of radioactively labelled P compounds. However, to the best of our knowledge, this has not yet been done, possibly because 32P and 33P are isobaric with the stable S isotopes 32S and 33S, respectively. Therefore, to trace the fate of labelled P, a soil sample must be free of S, since a simple subtraction of images without [showing the natural abundance of 32S (95.02%) or 33S (0.75%)] and with P labelling will not be practicable due to the likely heterogeneous distribution of S in both sample images. Another possibility may be waiting for the decay of 32P or 33P and measuring the spatial distribution of the decay products (32S or 33S), and superimposing the natural isotopic S distribution in the soil sample. The higher secondary ion yield of S isotopes compared to P may be an additional benefit from this approach. To advance this approach requires tracer concentrations and blind correction factors, further methodological test whether such small changes in the abundance of 32S or 33S can be detected as well as a systematic assessment of the possible effects of radioactive decay on the binding and fate of the tracer.
In summary, the capabilities of NanoSIMS provide a compelling argument for its application assimilatory processes in soil P research at the sub-micron level, especially when combined with complementary non-destructive, spatially-resolved speciation techniques such as P µ-XANES spectroscopy (cf. chapter 3.6).
3.6 Synchrotron-based X-ray spectroscopy
Since the early 1990s the use of synchrotron radiation (electromagnetic radiation produced in a synchrotron) has enabled the development and application of several X-ray absorption based spectroscopic and microscopic analytical techniques for P speciation and molecular-scale processes in soil and related samples. In the last decade, this has been promoted further by the rapid development of the number and capabilities of beamlines, which are, at least partly, dedicated to those types of samples. Essentially, this development is linked with the high brightness and intensity and wide tuneability of X-ray beams using synchrotron radiation. Therefore, a higher sensitivity is achieved compared to conventional X-ray sources (e.g., X-ray tubes). Most of the X-ray methods are based on the photo-electric effect. Briefly, X-ray photons get absorbed by the target element, thus promoting its core electrons (e.g., from K- or L2,3 shells) to higher energy levels or emitting photo and auger electrons into the continuum. Below the element-specific binding energy (BE) of the core electron, the absorption is small; on the contrary, at energies close to the BE of the core electron, a sharp increase in the absorption occurs. However, a further increase in energy decreases the probability for photo absorption again, leading to an edge-like structure in the absorption coefficient; this is the absorption edge, sometimes also called “white line”. As a consequence, the absorber atom is left in an unstable exited state. To return to the ground state, the vacancy (core hole) is rapidly filled by an electron from a higher energy level and the difference in both energy levels is emitted in the form of characteristic fluorescence photons and/or as auger-electrons. There are three main techniques utilizing the fact that absorption energy, energy of emitted electrons and fluorescence photons are characteristic for an element and its electron configuration: (1) X-ray fluorescence (XRF) spectroscopy, (2) X-ray absorption spectroscopy (XAS), and (3) X-ray photoelectron spectroscopy (XPS). For in-depth explanations of the theory and instrumental setup of XAS, XPS, and XRF we refer to the pertinent text books (e.g., XAS: Stöhr, 2003; Calvin, 2013; XPS: Briggs and Seah, 1990; XRF: Wobrauschek, 2007).
3.6.1 Fluorescence (XRF) spectroscopy
Energy dispersive X-ray fluorescence (XRF) spectroscopy is a fast, non-destructive technique enabling quantitative, but no qualitative, multi-element analyses of environmental samples (Bamford et al., 2004). Although XRF spectroscopy can be carried out with laboratory-based or even handheld X-ray tubes, the utilization of synchrotron radiation as the excitation source is becoming more popular; this is because higher quality (better signal-to-noise-radio) and more accurate spectra can be recorded as a result of reduced detection limits (Sarret et al., 2013). This is even more beneficial for soils in which sample matrix effects, causing high background noise levels and reduced sensitivity, constrain the use of XRF (Mukhtar and Haswell, 1991). XRF spectroscopy is commonly used to determine the total P concentration in environmental samples, such as soil (e.g., Baldwin, 1996; Baranowski et al., 2002; Morgenstern et al., 2010) and plant materials (e.g., Reidinger et al., 2012). Concentrations obtained are comparable with results from common digestion-based techniques or ICP-AES and ICP-MS. Nevertheless, XRF spectroscopy is often the preferred method due to the ease of sample preparation, and the relatively rapid sample through-put, thus enabling a more cost effective screening of very large sample sets, even in the field (e.g., Bamford et al., 2004).
Synchotron based total X-ray fluorescence (TXRF) spectroscopy is one of the many varieties of XRF, capable of determining elemental concentrations in the ultra-trace range; indeed, measurements of Pt atmospheric aerosol particulates are possible at concentrations as low as 0.2 to 0.3 ng P m−3 (Fittschen et al., 2013). This is achieved by decreasing the incidence angle of the incoming beam below the critical angle of total reflection; this minimizes penetration depth, absorption and scattering of the incoming beam in the sample matrix and, thus, reduces background noise and increases sensitivity (Wobrauschek, 2007). Liquid samples are best suited for TXRF spectroscopy (Wobrauschek, 2007), and have been applied for P analysis in soil extracts (e.g., von Bohlen et al., 2003; Hoefler et al., 2006), and plants (Antoine et al., 2012). In a recent study by Towett et al. (2013), TXRF spectroscopy of solid powdered soil samples yielded P concentrations comparable to results obtained by ICP-MS, without a need of a labour-intensive digestion as required for ICP-MS.
3.6.2 X-ray absorption spectroscopy (XAS)
X-ray absorption spectroscopy is based on measuring the variation of the absorption coefficient of a sample as a function of the applied X-ray energy in order to obtain an absorption spectrum. This can be done either directly or indirectly. The direct method monitors the intensity of the beam transmitted through the sample (transmission mode). Indirect methods include monitoring the emitted total or partial fluorescence yield (FLY), or the sample drain current as total (TEY) or partial electron yield (PEY). Generally, spectra obtained by these different detection modes are comparable but differ in their analytical depth; TEY and PEY measure the surface of the samples whereas FLY is rather a bulk method (P L2,3-edge: TEY ≈ 5 nm vs. FLY, ≈ 70 nm: P K-edge TEY ≈ 20 nm vs. FLY, ≈ 200 nm) (Kruse et al., 2009). For soil P analyses, FLY is often more valuable, since the lower background of the FLY signal results in a better signal-to-background ratio and spectra are not affected by surface charging as often observed for TEY (Stöhr, 1992). Conversely, it should be noted that spectra recorded in FLY mode for highly concentrated and thick samples can be distorted by self-absorption effects (e.g., Shober et al., 2006; Toor et al., 2006). Essentially, these effects are attributed to a reduction in the penetration depth, causing re-absorption of the fluorescence photons; this is not an issue for spectra recorded in TEY mode. Therefore, samples with high P concentrations should be diluted or analyzed using the TEY detection mode. However, it should be noted that for samples where surface structure is different from that of the bulk material, TEY and FLY spectra may differ in their spectral features; this is a direct consequence of the much shallower sampling depth of TEY (see above) (e.g., shown for vivianit in Toor et al., 2006). Furthermore, due to the generally low penetration depth (a few microns) of the X-rays used for P XAS measurements, data collection in transmission mode is often impractical. The relatively low X-ray energy used for collecting P XAS spectra requires measurements under high vacuum conditions (P K- & L2,3-edges) or under ambient pressure in He-purged (P K-edge) chambers to minimize attenuation effects by the air. The latter condition also enables solid-state in-situ measurements of moist or liquid samples (Kelly et al., 2008); whereas solid samples must be dried in advance for measurements under vacuum. Liquids must be inserted into a liquid cell (Rouff et al., 2009) that seals the liquid from the vacuum by an appropriate P-free thin film (e.g., ultralene). One of the main advantages of XAS is the relatively straightforward preparation, simply by powdering and spreading a few milligrams as a thin film on a double adhesive tape which is attached to a sample holder. An example of a P XAS spectra recorded at the P K-edge (i.e., electrons from the K-shell or 1s are exited) and L2,3-edges are shown in Fig. 9a,b.

(a) Stacked and normalized P K-XANES fluorescence yield spectra of selected soil-relevant P reference compounds and (b) the respective P L2,3-edge spectra. (c) Long-range scan of amorphous Ca-phosphate at the P K-edge visualizing the XANES and the EXAFS region of the spectrum in energy space, (d) respective k-space transformed, and (e) Fourier transformed χ(k) data of amorphous Ca-phosphate, giving information on the type and distances of bondings (Kruse, 2013, unpublished).
The marked increase in intensity around 2,150 eV corresponds to the P K-edge. Based on this edge, the spectrum is conventionally divided into the X-ray Absorption Near Edge Structure (XANES, synonymous with Near-Edge X-ray Absorption Fine Structure: NEXAFS) region extending from approx. ± 50 eV of the edge and the remaining Extended X-ray Absorption Fine Structure (EXAFS) region, extending from around 600 to 1,000 eV above the edge. The exact ranges depend on the edge probed (Fig. 9c). The acronyms also provide the names given to the respective XAS techniques (i.e., P XANES and P EXAFS, preceeded by the edge which is probed; e.g., P K-edge). The XANES region, characterized by relatively intense features, is sensitive to the P oxidation state and local environment of the P atom; i.e., bond angle, type, and geometry of coordinating cations. The EXAFS region, the oscillating part of the XAS spectrum resulting from interference between the outgoing and backscattered electron waves, provides local information about P inter-atomic bond distances as well as coordination neighbors and numbers with the absorbing atom (e.g., Stöhr, 2003). For EXAFS, the spectra are mathematically transformed from energy-space (Fig. 9c) to the k-space (Fig. 9d). The corresponding amplitude of the Fourier transform χ(k) is shown in Fig. 9c. The experimental curves shown in Fig. 9d and 9e are fitted to a theoretical model, simulating the individual scattering pathways. The first peak in the Fourier Transform (Fig. 9e) corresponds to the first nearest neighboring shells (P–O), the second peak is due to multiple scattering paths followed by peaks from P–Ca bonds. The capabilities of P EXAFS are restricted by the generally weak EXAFS oscillations produced by elements with low atomic number (1 to 20) such as P as compared to elements with high atomic number. In the case of soil P analyses, this is even more challenging as the ubiquitous presence of naturally occurring sulfur (S) can distort the P EXAFS spectra by superimposing spectral features originating from the S K- or L2,3-edges appearing only approx. +325 eV and approx. +25 eV above the respective P edges. This might explain why P EXAFS has only been used so far to track changes in P bonding in defined and S-free artificial samples (Rose et al., 1997; Rouff et al., 2009). Recently, Abdala et al. studied P reactions at the goethite water/interface by P EXAFS and observed monodentate, and bidentate mono- and binuclear complexes (Dr. D. Abdala, personal communication).
The principal advantage of these two XAS techniques is the recording of element specific spectra that reflect the weighted sum of the local bonding of all P atoms within a sample. For soil samples, however, this makes subsequent spectra evaluation complicated because of the vast number of different P species and, thus, P bonds and chemical environments. Therefore, various deconvolution approaches have been applied to extract qualitative and quantitative P data from spectra of samples with unknown P composition. All of these methods are based on spectra of known Po and Pi reference compounds that are relevant to soil, and preferably recorded at the same beamline but sometimes also taken from the literature (e.g., Brandes et al., 2007; Kruse and Leinweber, 2008; Kruse et al., 2009; Ingall et al., 2011). Detailed descriptions of characteristic XANES spectral features of various Po and Pi reference compounds were compiled by Hesterberg (2010) for the P K-edge and by Kruse et al. (2009) for the P L2,3-edge. Similarly extensive spectra libraries for P EXAFS have not yet been published.
It is important to differentiate between the capability of XAS to distinguish between spectra of pure Po and Pi references compounds and to quantify those compounds also in real mixtures in soil. Initial indications of major P compounds in a sample can be provided by visual comparisons between sample and various reference spectra for the presence or absence of distinctive spectral features such as a pre-edge peak for Fe-associated P (see Fig. 9) or a post-edge shoulder for Ca-associated P. Mathematical deconvolution algorithms are commonly applied to determine the optimum combination of spectra of relevant P reference compounds for extracting quantitative information on individual P species. For spectral recording in FLY mode, it is important that the self-absorption effects are avoided by diluting the P standards in a P-free matrix (e.g., B-nitride, sea sand, liquid glass) or by applying mathematical algorithms for spectra correction (Ravel and Newville, 2005). Alternatively, the respective TEY spectra should be used; as discussed previously, unlike FLY spectra these are not affected by self-absorption. Linear combination fitting (LCF) is commonly used for spectra fitting, and is already included in various free XAS evaluation software packages, such as Athena (Ravel and Newville, 2005) and SixPack (Webb, 2005).
The most critical requirement for effective LCF analysis is the appropriate choice of a set of representative P reference compounds. Therefore, as well as visual inspection, the selection of P reference compounds should be based on all other available information relevant to the chemical nature of the sample (Kelly et al., 2008); e.g., the total elemental composition and stoichiometric mass balances, pH, and/or results of other complementary methods, such as 31P NMR, XRD or wet chemical analyses. Additionally, statistical analyses such as principle component analysis (PCA) and adjacent target transformation (TT) also have been used to determine the selection of relevant P reference compounds for subsequent LCF analysis (e.g., Beauchemin et al., 2003; Lombi et al., 2006; Seiter et al., 2008). However, the majority of P XANES studies, even those not having used PCA or TT for selection, used not more than four reference compounds for LCF; this appears critical given that several combinations of reference compounds can yield similar fitting results. Since the obtained fitting models cannot provide a perfect representation, fitting results must always be cross-checked for their plausibility (e.g., Giguet-Covex, 2013); this can be achieved, for example, by complementary analysis or stoichiometric calculations as opposed to relying solely on statistical goodness-of-fit parameters (χ2 or R-value). Systematic studies to directly validate and improve the precision and accuracy of XAS fitting results are urgently required, as performed for only simple binary mixtures by Ajiboye et al. (2007a, 2007b). For example, Eveborn et al. (2009) suggested the use of derivative spectra for fitting as the 1st derivative; it is likely that these spectra are less sensitive to variations in absolute intensities that are brought about by normalization or self-absorption. These approaches, however, have not yet been tested. Another new promising approach is to employ partial least squares regression (PLSr) models to deconvolute artificial mixtures of reference compounds; this would use artificially produced mathematical data sets for PLSr calibration of the XANES spectra (Siebers et al., 2012). Based on promising results obtained with Cd compounds, these researchers adopted this approach to evaluate P K-edge XANES spectra of bone char amended soils, and showed for the first time the formation of a Cd-P-phase by solid-state speciation (Siebers et al., 2013).
Regardless of the method used to deconvolute P XAS spectra, the differentiation and detection of individual P species and, therefore, the specificity of XAS, strongly rely on the presence of unique features in the spectra of P reference compounds. This dependency explains the limited specificity of P K-edge XANES spectroscopy, since many soil relevant P species lack clearly distinctive spectral features (e.g., Brandes et al., 2007; Kruse and Leinweber, 2008; Kruse et al., 2010) as compared to sulfur K-edge XANES (Fleet, 2005). This is not surprising, however, since the majority of P in soil involves P(V) in the PO4 tetrahedra. As such, the direct chemical environment around the P atom varies only very slightly and, therefore, only relatively small spectral variations are observed among different soil P species. As a rule of thumb, Beauchemin et al. (2003) postulated that a P species can be detected by P K-edge XANES if it represents 10 to 15% of Pt and has a spectrum that is unique from other P species. For instance, marked spectral features for Fe- and Al-associated Pi have been used to distinguish between P sorption by Fe and Al minerals in artificial mixtures of ferrihydrite and boehmite (Khare et al., 2004). Furthermore, peak assignment and spectral interpretation can be facilitated by molecular orbital calculations (cf. chapter 4.2) as undertook for P sorbed to Fe- and Al-(hydr)oxides (Khare et al., 2007).
Published work clearly indicates that P K-edge XANES spectroscopy is more suitable for speciating Pi species (e.g., Sato et al., 2005; Kruse et al., 2010; Ingall et al., 2011; Siebers et al., 2013) than to detect or distinguish different Po species; this is because most Po spectra are too similar to be separated from each other and from Pi compounds (Brandes et al., 2007; Kruse and Leinweber, 2008). It is suggested that this lack of sensitivity explains above all why previous P XANES studies (e.g., Beauchemin et al., 2003; Lombi et al., 2006; Güngör et al., 2007) failed to identify any Po species in soil and manure samples (cf. chapter 2). Indeed, complementary methods, such as wet cemical analyses or 31P NMR spectroscopy, clearly show that significant proportions of Po exist in such materials (e.g., Liu et al., 2013; 2014; Hashimoto et al., 2014). Phytic acid (myo-inositol hexakisphosphate), which is the predominant Po compound in soil (Harrison, 1987), has often been used as a proxy for Po in P K-edge XANES data evaluation (e.g., Shober et al., 2006; Ajiboye et al., 2008; Prietzel et al., 2013). This, however, neglects the fact that various metal phytates (e.g., Fe, Al, Ca) have different spectral features, e.g., an energy shift up to 1 eV, or the presence or absence of a pre-edge (He et al., 2007). Although still not investigated in detail, it is likely that phytic acid sorbed to Fe-, Al-, and Ca-minerals exhibit similar spectral variations (e.g., pre-edge peak, shoulder). Hence, this has to be taken into account during quantitative spectra evaluation in order to avoid misassignment and overestimation of Pi associated with Fe, Al, and Ca, since phytic acid is mostly stabilized and accumulated in soils via sorption to Fe-, Al-, and Ca-minerals (Celi and Barberies, 2005). Due to the inherent Po speciation difficulties, it is advisable to combine P K-edge XANES spectroscopy with complementary methods that are more sensitive to Po chemistry; e.g., 31P NMR, as reviewed in 3.1 (e.g., Ajiboye et al., 2007a, 2007b; Negassa et al., 2010; Liu et al., 2013; 2014; Hashimoto et al., 2014) and to cross evaluate the results for their plausibility.
A further technique to overcome the limitations of P K-edge XANES in Po speciation is the analysis of P L2,3-edge XANES spectra. The L2,3-edge spectra of various Po and Pi reference compounds are characterized by more spectral features than the corresponding K-edge spectra (Kruse et al., 2009), indicating enhanced specificity. Early examples of co-applying P K-edge and P L2,3-edge XANES were very promising (Schefe et al., 2009; Kruse et al., 2010; Negassa et al., 2010); these researchers demonstrated clearly that samples whose spectra were similar at the P K-edge displayed larger differences at the P L2,3-edge. However, despite these promising results, it should be noted that L2,3-edge XANES is still in its infancy. More widespread application of the technique in soil P speciation studies is still hindered by its much higher P concentration required to obtain a good spectrum compared to the K-edge (≈ 5 to 10 g P kg−1 vs. ≈ 0.1 to 0.3 g P kg−1); also, soil matrix related effects (non- linear background and charging effects) also continue to present limitations.
In summary, a full exploration of the far-reaching potential of X-ray absorption spectroscopy requires focused research in the following areas: improved methods for enriching P in the test samples; enhanced sensitivity of beamline endstations; circumvention of self-absorption problems, and of those arising from non-specific spectral features, and improved and validated data evaluation algorithms, including mathematical spectra deconvolution.
3.6.3 X-ray photoelectron spectroscopy (XPS)
X-ray photoelectron spectroscopy (XPS), also termed electron spectroscopy for chemical analysis (ESCA), is similarly based on the photoelectric effect as described above. However, in contrast to XAS, a monochromatic X-ray beam (e.g., Al Kα: hν = 1486.6 eV, or Mg Kα: hν = 1253.6 eV) is used for XPS to excite core electrons from a sample. The kinetic energies of all emitted electrons are then measured via an energy disperse detector, and the binding energy (BE) is calculated as BE = hν – KE – Φ – Ech (where hν is the photon energy, KE is the measured kinetic energy, Φ is the spectrometer work function, and Ech is the surface charge). The obtained BE is characteristic for a given element (e.g., P) and its various core level electrons (e.g., P 1s, 2p1/2, 2p3/2 …) and is also sensitive to its oxidation state and the local chemical environment surrounding the atom. Thus, XPS provides information on the element composition and ratio of the sample (e.g., P concentration). The oxidation state and variations in the P binding energy (i.e., chemical shifts) can then be used to differentiate P-species in the sample. In Table 1 chemical shifts of some P refrence compouds are complied.
| Binding energy | |||
|---|---|---|---|
| P compounds | P (2p3/2) | P (2s) | P (1s) |
| / eV | |||
| FePO4 | 133.75 | 191.05 | 2148.15 |
| Mn3(PO4)2 | 133.70 | 191.00 | 2148.00 |
| Ca3(PO4)2 | 132.90 | 190.20 | 2147.10 |
| Ca2P2O7 | 133.80 | – | – |
| Ca10(PO4)6F2 | 133.60 | – | – |
| Ca10(PO4)6(OH)2 | 133.80 | – | – |
| CaHPO4 | 133.80 | – | – |
| K2HPO4 | 132.80 | – | – |
| AlPO4 | 132.90 | 191.20 | |
By deconvoluting the XPS spectra into sub-peaks using Gaussian–Lorentzian functions, peak areas of individual P species can be calculated and converted into proportions. The sensitivity of XPS analysis can be enhanced when monochromatic synchrotron based X-ray sources are used for XPS. This also allows the measurement of diluted samples due to the much higher brilliances of the X-ray source. Furthermore, the possibility of tuning the photon energy of the X-ray beam enables the collection of XPS spectra as a function of photon energy. This can be beneficial since selected P spectral features can be enhanced due to resonance effects if the scanned photon energy range covers an absorption edge of P (e.g., K- or L-edges. This effect is used for so called high resolution resonant XPS (ResXPS) (e.g., Reinert and Hüfner, 2005), but has not yet been used in soil P studies. Nonetheless, high resolution ResXPS may be a valuable tool for the deconvolution of P peaks at the valence band region, and may improve the understanding of the origin of complex spectral features in P containing compounds.
It has to be noted that only electrons from the near surface can be captured by the detector due to the small penetration depth of the emitted photoelectron. Therefore, information gained from XPS is always restricted to the upper ≈ 10 to 15 nm of the sample surface. The analytical depth can be varied by angle dependent measurements, and depth profiles obtained by combinations with surface abrasion techniques. XPS experiments usually require ultra-high vacuum conditions to avoid any contamination of the surface by adsorbates from the ambient atmosphere. Besides vacuum stability and grinding, no other sample requirements or treatments are necessary. Samples are most of the time simply applied to a double-sided tape. However, recent advances in synchrotron-based XPS also allow measurements at ambient pressure and of hydrated samples (Yamamoto et al., 2007). Traditionally XPS, both lab- and synchrotron-based, has been applied mostly in material sciences (e.g., Lu et al., 2000, and references within) and has been less frequently used in soil science to investigate the composition of the surface layer of soil particles (biogeochemical interfaces) (e.g., Barr et al., 1999; Amelung et al., 2002). A few soil P-related XPS applications studied P sorption processes (e.g., Martin and Smart, 1987; Martin et al., 1988; Nooney et al., 1996; Mallet et al., 2013). For instance, Nooney et al. (1996) used shifts in P(2p) BE to track changes in the chemical state of P sorbed to Fe2O3 as a function of absorption time (up to 26 h). The observed increase in BE by ≈ 0.4 eV indicated that P became more oxidized with reaction time. To date, the majority of soil related XPS studies have used lab-based X-ray sources, most likely due both to the general limited access to beamlines and the fact that not all beamlines accept “dirty” soil samples. However, this is becoming less of a problem as beamlines capable of XPS are increasingly available and are prepared to accept soil samples. In view of the capabilities of XPS in elucidating the composition and heterogeneity of soil particle surfaces, it is likely that the increase in beamline accessibility will accelerate the application of synchrotron- based XPS to soil P studies.
3.6.4 Spatially resolved X-ray spectroscopy and combinations of techniques
Detailed information about the concentration and speciation of P in soils is obtained by the macro-scale, bulk spectroscopic techniques (XRF, XAS, XPS) described above. However, P heterogeneity in soils extends over a massive continuum from the nanometer to field scale. It is critical to obtain detailed information on spatial distribution and differences in P speciation at the so-called microsite scale (e.g., boundary surfaces of aggregates and pores as well as in hot-spots of P turnover such as the rhizosphere). Fundamentally, this microsite-scale heterogeneity controls important P biogeochemical cycles in soils, as proposed in the “microreactor concept” (Hesterberg et al., 2011). Processes in P cycling at this scale cannot be studied in bulk soil analyses, but they can be resolved using X-ray spectroscopy. Hence, spatially resolved X-ray spectroscopy (i.e., X-ray microscopy) is becoming more and more applied in soil P research, and its capabilities have advanced rapidly.
For X-ray microscopy the beam is focused on a small spot, and spatially resolved data are obtained by moving the beam spot to the area of interest (often called: µ-XANES, µ-EXAFS, µ-XRF). Additionally, a series of single images can be collected by recording the transmitted X-ray intensity or FLY for each scanned pixel as a function of sample position and energy. For the latter, the series of images can be aligned (stacked) to extract complete spectral information for each scanned pixel (e.g., Scanning Transmission X-ray Microscopy: STXM). In particular, if synchrotron radiation-based techniques are used, the spot size of the beam can be reduced drastically, enabling lateral resolution from micrometers down to < 50 nm without significant loss in sensitivity. It must be noted that mapping of a small region of sample at such a high spatial resolution requires more time than the respective bulk measurements, and the required sample treatment is less straightforward. In general, the sample surface should be flat to avoid spatial disorder due to sample morphology. This can be achieved, for instance, by first embedding the sample in epoxy resin and subsequent cutting into thin sections (e.g., Lombi and Susini, 2009). XAS measurements in transmission mode require very thin sections due to the low penetration depth of the beam, whereas thicker samples as prepared by drop coating (Brandes et al., 2007) can be measured in fluorescence mode.
Synchrotron-based, laterally resolved X-ray fluorescence (μ-XRF) microscopy is commonly applied for element mapping in environmental samples (Majumdar et al., 2012) and has been used frequently to map P in soil and sediment samples (e.g., Lombi et al., 2006; Brandes et al., 2007; Frisia et al., 2012). The use of energy dispersive detectors allows simultaneous mapping as well as quantification of, and spatial analysis with, other elements relevant to P cycling (e.g., Al, Fe, Na, Ca, Mg). It can be highly beneficial to combine µ-XRF and P µ-XANES, because knowledge about the spatial association of elements with P can help to select relevant reference standards for a subsequent fitting of P XANES spectra. Another benefit of a preceding µ-XRF-based P mapping is the clear identification of P hotspots in the sample. Focusing P µ-XANES measurements at these hotspots may yield XANES spectra with higher signal-to-noise ratios and less complexity compared to respective bulk analyses (dilution effects) and, thus, facillitate more precise P speciation and quantification. Despite these advantages and benefits, spatially resolved P X-ray spectroscopy is still a relatively new tool for soil P research compared to its use in the determination of other elements (C, Fe, etc.), and, as a consequence, only a few P studies have been published. To the best of our knowledge Lombi et al. (2006) were the first workers to use both bulk and combined µ-XRF and µ-XANES in order to elucidate the fate of different P fertilizers in soils. In calcareous soils, they found that Ca-phosphates seem to dominate P precipitation processes and that Al- and Fe-complexes were only of secondary importance. Others have used this combined technique to characterize P in marine sediments (Brandes et al., 2007), to study the influence of carboxylic acid addition on P sorption (Schefe et al., 2011), and to investigate changes in the soil P speciation during soil genesis (Giguet-Covex et al., 2013). However, some of these studies also point to an inherent drawback of spatially resolved techniques when applied to complex samples. Essentially, the large uncertainty in selecting a representative area of the sample for analysis presents a sizeable challenge to the up-scaling of the results. Most samples taken from soils are characterized by significant intra-sample heterogeneity; generally, this is reduced to some extent by bulk determinations. Since spatially resolved µ-analyses are relatively time consuming, and access to dedicated imaging beamlines can be limited, P µ-analyses are often restricted to a few spots/areas of the sample (P-hotspots or subjective choices of the investigator). As a result, differences in P speciation between soil treatments are often very difficult to identify and to distinguish from natural soil P heterogeneity at the µ-scale (Lombi et al., 2006). As such, complementary bulk measurements are urgently required (Schefe et al., 2011).
In summary, synchrotron-based X-ray spectroscopy offers unique possibilities in the speciation of soil P and in the use of this information to deepen our insight into the soil P cycle. In order to realize its potential, more holistic (bulk and spatially resolved) and combined (see, e.g., complementary techniques in this review) approaches are required.
4 Methods for assessing soil P reactions
4.1 Sorption isotherms
Sorption isotherms describe the adsorption of a material by or desorption from the soil solid phase as a function of the material's equilibrium concentration in solution at constant temperature and pressure. The common approach to obtain P sorption isotherms involves the interaction of a soil sample with solutions containing a range of concentrations of P—usually as KH2PO4 in a 0.01 M CaCl2 matrix or as close as possible to the ionic background composition of natural conditions—for a given time in a reaction vessel. The choice of reaction (contact) time is important, since it is known that P adsorption in soils is a continuum between initial, rapid adsorption at high affinity mineral surface sites and slow adsorption of P due to gradual diffusion into aggregates or precipitation of P minerals. Usually, the initial, rapid reaction is investigated by isotherms. Therefore, the contact time must be sufficiently long to reach equilibrium but also short enough to prevent significant amounts of P from undergoing the subsequent slow retention process, e.g., 24 h (Barrow, 1978).
Following the equilibration reaction, the samples are filtered and the concentration of P remaining in the filtrate is determined. In the main, there are two different experimental designs for these laboratory-based determinations of P sorption isotherms: static close batch and continuous open flow. In soil P research, the former is by far the most conventionally applied approach (Limousin et al., 2007), and the latter is only seldomly applied.
Conventional batch experiments are inexpensive and simple techniques, requiring only the stirring or agitation of the soil sample with the P solutions in a reactive vessel (e.g., centrifuge tube). The solid/solution ratio is generally advised to be between 1:2 to 1:4 (w:v) (Porro et al., 2000). However, it is difficult to simulate solid/solution ratios of natural systems, since the ratio for suspended particles is often too low and that for soil is too high (Limousin et al., 2007). Furthermore, very slow reactions are difficult to assess as long periods of shaking can lead to the destruction of soil particles (Sposito, 1984).
In continuous open-flow systems, the solution is added to the soil sample (≈ 0.5 to 2 g) at a constant rate, with the soil positioned either as a dry sample or suspension on a membrane filter in the flow reactor. The thin soil disk is then flushed (saturated or unsaturated conditions) with the solution by connecting the filter holder to a fraction collector and a peristaltic pump. The solution continuously injected into the flow reactor is originally free of P. There are different ways to inject P into the reactor: (1) instantaneously injecting at the inlet (“direct” injection), (2) continuously injecting during a time period (finite step injection), or (3) continuously injecting after a defined time (infinite step injection) (Limousin et al., 2007). By adjusting the flow rate, the mean residence time can be chosen; compared with the closed batch method, this makes it easier to investigate the kinetic dependency of P sorption (Sparks, 1985). After defined time intervals the effluent can be collected. When the concentration of P in the effluent is equal to the concentration in the eluate, the equilibrium is reached. A disadvantage of this approach is the fact that the higher solid/solution ratio usually requires longer contact times to reach equilibrium compared to the batch method (Qualls and Haines, 1992). During either method, microbial P fixation may overestimate P sorption, especially during long contact periods in P depleted soils; this can be suppressed by the addition of HgCl2, or, even more effectively, by gamma-radiative sterilization. However, for the later it has to be noted that gamma-sterilization can also affect soption properties of the soil (e.g., Bank et al., 2008) which is also true for autoclaving (Serrasolses et al., 2008).
In the continuous open-flow system, released competitive anions are flushed out by the repeated renewal of the solution, whereas in a closed batch system the solution becomes relatively enriched with these anions which, therefore, progressively compete with P for adsorption sites. As a consequence, adsorption should be higher in the open than in the closed batch systems. While studies directly comparing P adsorption between the two methods are still rare, those using sorbates other than P report the opposite effect and generally attribute this to (1) the presence of immobile water in the soil sample, (2) solid/solution ratio effects, or (3) the non-achievement of chemical equilibrium due to significantly lower residence times than the mean reaction time (Limousin et al., 2007). Nonetheless, the few studies that have compared the two methods with respect to P adsorption in soils indicate that batch-generated P adsorption isotherms likely underestimate the extent of P adsorption compared to continuous flow-based methods (Miller et al., 1989).
The batch approach can also be applied to obtain P desorption isotherms; this is performed, for example, by adding a P-free solution, usually in 0.01 M CaCl2 (or as close as possible to the ionic background composition of natural conditions) to the soil samples at various soil solution ratios for a specified shaking time (e.g., Sharpley, 1985). Another way is to use an extraction solutions with fixed soil:solution ratio and shaking time, but sequential extraction using solely the extraction solution (e.g., Bhatti and Comerford, 2002) or the extraction solution combined with an anion exchange resin (Bache and Ireland, 1980; Yang and Skogley, 1992; Sato and Comerford, 2006) until the P desorption is exhausted or a pattern of P-release is established. For continuous flow experiments the strength of the extraction solution can either be continuously increased or kept constant with varying flushing levels and, thus, varying soil:solution ratios. The P released into the solution (desorbed) is measured when it reaches equilibrium with the adsorbed phase (batch) or after defined time intervals (continuous flow). The time needed to reach desorption equilibrium can be much longer than that required for adsorption (Tisdale et al., 1985). This can be misinterpreted as irreversible P adsorption, particularly where short desorption steps are used and the system has not reached the equilibrium (pseudo-hysteresis). Lookman et al. (1995) showed that this phenomenon is due to the much slower P desorption kinetics caused by the so called “ageing effect”, i.e., progressive diffusion of the sorbate from the surface into the soil particle, formation of sorbate inner-sphere surface complexes, and the crystallization of new solid sorbate phases (Strawn and Sparks, 1999).
Data obtained from sorption experiments are usually mathematically fitted to empirical equilibrium isotherm equations; these concisely summarize the data and provide correlative indices with soil properties that may aid our understanding of the sorption process. A comprehensive overview of empirical equations can be found in, e.g., Barrow (1978) and Goldberg (2005). The two most common empirical equations applied to fit P ad- and desorption in soils are (1) the Freundlich isotherm (Freundlich, 1909), and (2) the Langmuir isotherm (Langmuir, 1918), or modifications of them. Both are characterized by a concave curve (“L”-shape) (Giles et al., 1974) that reflects the decreasing ratio of adsorbed to solution P at equilibrium with increasing P addition. The "L" isotherms are generally divided into two subgroups: (1) the solid has unlimited sorption capacity (no strict asymptotic plateau is reached), and (2) the solid has limited sorption capacity (a strict asymptotic plateau is reached).
 ()
()with Kf representing the proportionality constant (mg kg−1) and n (dimensionless) denoting the empirical coefficient related to the binding energy of soil solids for P. According to this equation, the Freundlich isotherm does not reach a plateau at high P concentrations.
 ()
()where X is the amount of P adsorbed, C the P concentration in the solution, Kl the affinity (binding energy) coefficient (L mg−1 P), and Xmax is the adsorption maximum (mg P kg−1) at the plateau of the isotherm. The possibility to derive information about maximum P capacity can be viewed as an advantage of the Langmuir over the Freundlich isotherm equation. However, as already stated by Barrow (1978), this equation only describes adsorption over a limited range of concentrations, and Xmax calculated from observations at low concentrations often is exceeded at higher concentrations. Furthermore, the assumption of a constant affinity for P adsorption that is independent of the adsorbed quantity may oversimplify the absorption process, in which the affinity is a function of the amount of P adsorbed. Therefore, more complicated multi-surface Langmuir-type equations should also be applied when appropriate; e.g., Holford et al. (1974) and Hussain et al. (2012) partly consider this fact. However, the better fits are usually obtained with multi-surface Langmuir-type equations that can, at least, be partly attributed to the increased number of adjustable parameters (Mead, 1981; Goldberg, 2005).
Whereas in most P sorption studies Pi was used as sorbent, there are also a number of studies published using both Pi and Po (e.g., Berg and Joern, 2006) or Po as sorbent. In the case for the latter, Yan et al. (2014) recently used the Langmuir isotherm equation to model the sorption and desorption characteristics of various Po compounds (i.e., glycerophosphate, glucose-6-phosphate, adenosine triphosphate, myo-inositol hexakisphosphate) on three Al-(oxyhydr)oxides with different crystallinity (amorphous Al(OH)3, boehmite and α-Al2O3). They showed that the molecular-chemical structure, as well as the crystallinity of the Al-(oxyhydr)oxides, were the key factors affecting the sorption of the tested Po compounds. Similar sorption characteristic for various Po compounds were reported by e.g., Ognalaga et al. (1994) and Ruttenberg and Sulak (2011) for Fe-(oxyhydr)oxides (ferrihydrite, goethite, and hematite). In most cases, P sorption and complexation processes have been studied in single sorbate and pure mineral systems (e.g., Shang et al., 1996; Wang et al., 2013); however, there is a growing number of studies that evaluate the effect of competitive interactions with other sorbats (e.g., glyphosate, Cd, dissolved organic matter, amino acids) in the soil solution on P sorption either by pure Al- and Fe-minerals (e.g., Wang and Xing, 2004; Borggaard et al., 2005; Waiman et al., 2013) or within soil samples (e.g., Guppy et al., 2005; Negassa et al., 2008; Borggaard and Gimsing, 2008). Increasingly, studies are also evaluating in detail the sorption of P in soils from different sources (e.g., Shafqat and Pierzynski, 2014).
In summary, isotherms provide a powerful tool for describing P sorption and desorption characteristics of soils, especially if combined with complementary quantum-chemical modelling (e.g., Xuemei et al., 2012), IR or NMR spectroscopy in order to monitor the evolution of the adsorbed phosphate complexes as a function of, e.g., loading, reaction time, pH or presence of other sorbats (e.g., Arai and Sparks 2001; Luengo et al., 2006; Elzinga and Sparks 2013; Elzinga et al., 2013; Li et al., 2013b). Given that soils are highly dynamic and open systems (Sparks, 1985), the continuous open flow-based approach should be preferred wherever possible for describing P sorption in soils, since it is superior to the widely used batch-based approach in mimicking soil conditions (e.g., higher soil/solution ratio, dynamic flow). However, since P isotherms are not a thermodynamically inherent soil property, the experimental conditions during the isotherm measurement (e.g., experimental design, solid/solution ratio, electrolyte concentration, time of equilibration, range of initial P concentration, rate and type of shaking, and temperature) must always be provided alongside the results. Furthermore, it has to be kept in mind that the application of isotherm equations to experimental sorption data is basically only a curve-fitting procedure, and, therefore, the equation parameters are only valid for the experimental conditions under which they were obtained. Furthermore, before any equation parameters are used to describe P sorption processes or provide information on soil properties, their chemical meaning must be independently validated by elaborated sorption experiments (Goldberg, 2005).
4.2 Quantum-chemical modeling
During recent decades, simulations at the molecular level have been developed to provide an indispensable tool in Chemistry and related cross-disciplinary areas like Chemical Physics and Biochemistry. Starting from simple gas phase reactions, and successively through solution phase studies (e.g., solvation dynamics) and conformational dynamics of peptides to surface science topics such as catalysis, almost all areas of Chemistry are seen to benefit from the ever increasing sophistication of numerical algorithms and computer hardware. Among the few topics that have not received much attention so far are biogeochemical processes occurring in soil, and especially those related to the physicochemical behavior of P. At first sight this appears to be surprising since theoretical simulations of elementary processes in soil hold the promise to deliver a molecular level understanding. However, a straightforward implementation is hampered by the considerable complexity of the problem, with the soil being an extremely heterogeneous natural entity made up of, e.g., inorganic minerals such as clay and oxide particles, organic matter, soil solution and gases, with the details differing from site to site. Here, a possible solution arises from the comparison with experiments using prepared and, in terms of composition, well-characterized samples (e.g., Ahmed et al., 2012; 2014).
There is a hierarchy of computational methods for atomistic modeling (Cramer, 2004). On the simplest level, the interaction between the charged nuclei and electrons are described by empirical, so-called molecular mechanics (MM) force fields. These force fields are usually parameterized against macroscopic properties of the considered ensemble and hence implicitly include also quantum-mechanical effects. This has to be contrasted with explicit quantum-mechanical methods, the most prominent one in terms of the trade-off between efficiency and accuracy being density functional theory (DFT) (Koch and Holthausen, 2001). Central to DFT is the quantum-mechanical electron density (the nuclei are treated in classical approximation), calculated in terms of the so-called Kohn–Sham molecular orbitals; this connects to the chemist's view on molecular electronic structure. The flexibility of the molecular orbitals is provided in terms of an expansion into atomic orbitals. Although in principle an exact approach, in practice the quality of the DFT results depends on the so-called exchange-correlation functional. For the latter a plethora of examples exists, some developed for special purposes and others being more general. In the latter class falls the so-called Becke, three-parameter, Lee–Yang–Parr (B3LYP) functional that has also been used for P-simulations. Often accurate quantum-chemical modeling is necessary, e.g., for reactive sites only. This suggests a combination of quantum and empirical methods to describe the reactive site and the surrounding medium, respectively. For this purpose various blends of quantum mechanics/molecular mechanics (QM/MM) methods have been developed (Senn and Thiel, 2009). On a simpler level the surroundings can also be described as a classical dielectric medium, being polarized by the electron density in the quantum region and acting back on it. Among the more popular models is the so-called PCM (polarizable continuum model) (Tomasi et al., 2005). A further distinction has to be made concerning the treatment of surfaces, which can be either in terms of a finite cluster or using periodic boundary conditions. Examples of where quantum-chemical modeling can be applied to the interaction of phosphates with soil constituents are summarized as follows.
A central question in P research concerns the binding of phosphates to mineral surfaces. This issue has been addressed especially by Kubicki and co-workers in a number of papers. In an early work (Kwon and Kubicki, 2004) the pH dependence of phosphate binding to Fe-hydroxides, regarded as a small cluster of two edge-sharing Fe-octahedra (mimicking goethite, α-FeOOH) including a few explicit water molecules, has been described using DFT/B3LYP. Focusing on the asymmetric phosphate stretching vibration in the IR spectrum (cf. chapter 3.2), measured by Arai and Sparks (2001), as well as PCM-based reaction energies, it was concluded that the prevailing motif at low pH is a bi-protonated bidentate species, whereas at neutral pH, a mono-protonated monodentate inner-sphere complex is dominant. This tendency was confirmed in a comparative study of surface complexes of various oxyanions, including P adsorption on goethite, in Kubicki et al. (2007), in which emphasis has been placed on the correlation with IR and EXAFS data. The model was later extended to the binding of glyphosate, which showed monodentate complexes at all pH ranges (Tribe et al., 2006). The interaction of phosphate with a similar cluster model of boehmite (γ-AlOOH) was investigated by Li et al. (2010), who also calculated NMR chemical shifts and shielding anisotropies. Here, bidentate complexes were found to be the major species at different protonation states. More recently, Kubicki et al. (2012) were among the first workers to use periodic boundary conditions in calculations to describe P adsorption on goethite surfaces. This approach yielded evidence for a dependence of the binding motif on the crystal face. This paper also contains a critical review on the use of vibrational frequencies of phosphates for the assignment of binding sites, pointing to the variety of observed frequencies for seemingly similar samples. This was taken as further proof of heterogeneity of the samples. Other systems studied by these researchers include corundum (α-Al2O3), where a combination of ATR/FTIR experiments and DFT cluster calculations suggested a mixture of non-protonated and mono-protonated bidentate complexes at pH 5 (Li et al., 2013b). This paper also includes a review on previously proposed mechanisms for phosphate sorption on Fe- and Al-(hydr)oxides), and binding of phosphates to aqueous uranium complexes (Kubicki et al., 2009).
Periodic DFT calculations were also applied by Belelli et al. (2014), both for goethite and Al-rich goethite systems. In addition to previously studied complexes, all possibilities of mono-/bidentate and mono-/binuclear motifs have been investigated at different protonation states. It was shown that the respective phosphate vibrational frequencies correlate with experiment, provided that various motifs and pH values are taken into account. The difficulty in using IR data to determine adsorption motifs has also been addressed by Khare et al. (2007). As an alternative, these authors used P K-edge XANES in combination with semi-empirical extended Hückel calculations of density of states for Fe- and Al-(hydr)oxides in the cluster approach. Here it was concluded that phosphate adsorbs on ferrihydrite at pH 6 as a bidentate-binuclear complex. Particular attention to the effect of surface charge distributions for phosphate adsorption on a goethite dimer in the presence of a water layer was given by Rahnemaie et al. (2007). These surface charges were taken from empirical fitting of DFT data onto a bond valance model and corrected for dipolar interactions. They allowed simulation of phosphate adsorption (presence of non-protonated bidentate and protonated monodentate) and proton co-adsorption data. A detailed analysis of competitive water and phosphate binding to a Fe-(hydr)oxide cluster in terms of the electron density was performed by Acelas et al. (2013), providing insight into the role of the H–O...H hydrogen bonds. DFT cluster calculations have also been employed to study competitive sorption of, e.g., Cu(II) and phosphates on γ-Al2O3. Here, at pH 5.5, Cu(II)-phosphate surface complexes were found to be in the B ternary form, where the phosphate bridges Cu(II) and γ-Al2O3 in an outer sphere motif (Ren et al., 2012).
The research group of García-Mina has put forward a series of combined experimental and theoretical studies on soluble phosphate–metal–humic (P–M–H) substances, aiming to explore phosphate fixation by metal-humic complexes. The competition between P–M–H and M–H complexes was studied by molecular modeling for M=Al(III), Fe(III), and Zn(II) and H=salicylic acid (Guardado et al., 2008). Here, a combination of MM and semiempirical (PM3) modeling in the gas phase concluded that the stronger the P–M–H binding the weaker the M–H binding. This finding was taken as an explanation for the relatively low proportion of M–H complexes that are involved in P fixation. Subsequently, this study was extended to the DFT level focusing on M=Al(III), Fe(III), and Ca(II) (Urrutia et al., 2013). In another work, organic complexed superphosphates were studied by DFT calculations of Gibbs free energies using penta- and hexacoordinated Ca(II) complexes formed with phosphate and salicylate ligands (Erro et al., 2012). The pentacoordinated complex was found to be the most stable one. It was concluded that this is the structure involved in the reaction of the superphosphate, although the difference from the hexacoordinated species of about 2 kcal mol−1 appears to be negligible given the accuracy of the model. Experiments and calculations on 31P NMR chemical shifts have provided evidence for simultaneous formation of phosphate- and sulfate-Ca-humate complexes, the latter being the dominant form (Baigorri et al., 2013).
In summary, quantum-chemical modeling is emerging as a powerful tool for detailed investigations of binding motifs and reaction mechanisms of P-species in soil. In spite of its dominant role in other areas, atomic-level simulation has still not reached its full potential in soil science. The examples given provide evidence that substantial progress will come only from the combination of theory and experimentation using well-prepared samples (e.g., Ahmed et al., 2012; 2014).
4.3 Microbial biomass P and microbial P turnover
Although the microbial biomass contains usually only 0.4 to 2.5% of Pt in cropped soils and up to 7.5% in grassland soils (Oberson and Joner, 2005), soil microbial biomass P (Pmic) stores an essential labile P source for the vegetation (Richardson and Simpson, 2011). It can be a good indicator of P availability for plants (Sugito et al., 2010) and is composed of nucleic acids (75%), acid-soluble P-esters (20%), and phospholipids (5%) (Webley and Jones, 1971). Based on a meta-analysis, Cleveland and Liptzin (2007) derived a global soil microbial C:N:P ratio of about 60:7:1.
Most estimates of Pmic are calculated as the difference in P between extracts from soil taken before and after microbial cell lysis by biocides (Brookes et al. 1982, Hedley et al., 1982). The most common method is the fumigation extraction (FE) method by Brookes et al. (1982). In this method chloroform (fumigation) is used as a biocide and P is extracted from the soil samples by 0.5 M NaHCO3. The P concentrations are determined spectrophotometrically or by ICP. Since only a fraction of Pmic is recovered by the extraction (37%: Hedley et al., 1982; 40%: Brookes et al., 1982) conversion factors (Kp) of 0.37 or 0.4, respectively, are used to calculate Pmic. Although this method was developed and tested originally for neutral to alkaline soils (Brookes et al., 1982; Hedley et al., 1982) it has been applied for a wider range of soils (e.g., Joergensen et al., 1995; Blackwell et al., 2009). The impact of the soil-specific P-sorption on the result of this method was alleviated after modifications by Morel et al. (1996) using a shorter fumigation period (2h 15 min) followed by a longer equilibration period (40 h) and correction of measured values using a P sorption curve. For acidic soils (pH < 6.0) and soils with high P-sorption capacities, the method was modified by using 0.03 M NH4F/0.025 M HCl (Bray-1 solution) as the extractant instead of 0.5 M NaHCO3 (Oberson et al., 1997; Wu et al., 2000; Chen and He, 2004) or by using an anion exchange membrane method (Kouno et al., 1995).
The important role of phosphate-solubilizing microbes in soils has been recently reviewed by Kahn and Williams (1977) and Sharma et al. (2013). Microbially-mediated enzymatic hydrolyses transform Po into plant available P compounds. Phosphomonoesterates and phosphodiesterases are the most commonly investigated soil phosphatases. They catalyze the hydrolysis of phosphate mono- and diester bonds (e.g., Zalatan and Herschlag, 2006). Soil phosphatases occur as extra- and intracellular enzymes and are divided into acid and alkaline phosphatases according to their pH optima of activity (4 to 6 and 9.4 to 10, respectively). In general, acid phosphatases are believed to originate mainly from fungi and plants, whereas alkaline phosphatases originate from microorganisms only (Gianinazzi-Pearson and Gianinazzi, 1978). The role of phosphatases in soil has been reviewed, e.g., by Nannipieri et al. (2011). The activity of phosphatases is affected by soil properties, abundance and interactions of soil organisms, plant cover, leachate inputs, and the presence of various inhibitors and activators (Speir and Ross, 1978); it is generally negatively related to P availability (Sinsabaugh et al., 2008).
Phosphatase activity can be determined by quantifying either phosphate (produced by the mineralization of natural organic phosphate esters) or organic components after mineralization of artificial Po substrates over time (Samuel et al., 2010). A broad range of spectrophotometric methods has been developed to analyze the activities of diverse groups of phosphatases (e.g., phosphomonoesterases: Tabatabai and Bremner, 1969; phosphodiesterases: Browman and Tabatabai, 1978). However, they are all based on the addition of an artificial P substrate (e.g., p-nitrophenylphosphate) to a defined soil volume. The sample is then incubated for a defined period (e.g., 1 h) at an optimized pH (e.g., pH 6.5 for acid and pH 11 for alkaline phosphatases) by buffer-addition and an optimized temperature (e.g., 37°C). The concentration of the product released from the added substrate is determined colorimetrically (Tabatabai and Bremner, 1969) or fluorimetrically (Marx et al., 2001), which is proportional to the potential phosphatase activity at defined conditions. However, in soil samples low in enzyme activity and high in OM content (e.g., peat soils, sludge, organic layers), the DOM can cause high background absorbances in colorimetric assays, and quenching interference in fluorimetric assays. Information on the optimization of hydrolytic and oxidative enzyme methods for ecosystem studies was provided by German et al. (2011). Furthermore, Freeman et al. (2011) developed an approach based on HPLC to improve the quantification of enzymatic hydrolysis products.
Recently, a new method, soil zymography, has been developed for measuring the spatial distribution of exoenzyme activity, including phosphatases, at high resolution (Spohn et al., 2013b). This method can map the distribution of alkaline and acid phosphatases in soil (Fig. 10), but it is also suitable for the analysis of other hydrolases, in contrast to a similar method published by Grierson and Comerford (2006). The zymographic maps indicate that microbes and plants mineralize Po in different parts of the rhizosphere, signaling a potential for reduced competition between microbes and plant for P (Spohn and Kuzyakov, 2013a).

Distribution of acid and alkaline phosphatase activity in the rhizosphere of Lupinus albus analyzed by soil zymography. The calibration line is given at the bottom (Spohn and Kuzyakov, 2013a, reploted with permission from Elsevier). A color image is in the digital version of this article.
In summary, Pmic and the activities of phosphatases are valuable biochemical indicators of soil P status. The ratio of Pmic to Pt indicates the site-specific significance of Pmic in the P cycling and is management affected (Aponte et al., 2010). Activities of phosphatases indicate the soil's potential to mineralize Po, which usually forms “hot spots” in the rhizosphere by the interaction of plant and microbes. The P fractions already made available to plants can then be traced by other complementary methods among which diffuse gradients in thin films (DGT) recently has received widest acceptance (cf. chapter 4.4).
4.4 Diffusive gradients in thin films (DGT)
Diffusive gradients in thin films (DGT) is a solute sampling technique for studying labile solute fractions in soils, sediments, and water bodies. Originally developed as a tool for limnology and marine science (Davison and Zhang, 1994), it is now increasingly used in soil P research. In soils, the technique can either be applied for analyzing bulk soil samples, e.g., for the estimation of P bioavailability to plants (Mason et al., 2010; Tandy et al., 2011; Six et al., 2012), or as a spatially resolved chemical imaging tool allowing researchers to assess the distribution of P around biogeochemical hotspots (Santner et al., 2012).
 ()
()Here, M is the mass of P bound by the resin gel, usually measured in 0.25 mol L−1 H2SO4 eluates of the gel discs by the molybdenum blue method, Δg is the thickness of the diffusion layer, i.e., the sum of the diffusive gel and the membrane, D is the phosphate diffusion coefficient in the diffusion layer (provided by DGT Research Ltd.), A is the sampling area, and t is the sampling time. Assuming a limit of quantification (LOQ; usually determined as mean blank signal + 10 SD of the blank) of 10 µg L−1 for the molybdenum blue method and an eluate volume of 0.5 mL, the LOQ for the DGT application would be ≈ 0.35 µg L−1 P for a DGT deployment time of 24 h at 20°C.
For DGT measurements, sufficient soil moisture is required to ensure good contact between sampler and soil—both for maintaining continuous P diffusion into the sampler and ensuring comparability between tested soils. Therefore, bulk soil samples are wetted close to saturation and the paste is smeared onto the DGT sampler. Alternatively, the sampler can be pushed directly into soil of elevated water content, e.g., into a planted pot for in situ-measurements. Although arbitrary, 24 h is often chosen as the sampling time for soils (Mason et al., 2010). During sampling, P diffuses into the sampler and rapidly becomes depleted in the soil solution. This decline in P concentration is compensated by P desorption from the soil surfaces. Since P desorption from soil is usually not fast enough to maintain a consistent P concentration in the solution surrounding the sampler, a continuously extending P depletion zone adjacent to the DGT device is established. As a consequence, the P flux into the sampler, which is driven by the concentration gradient through the gel layer, progressively decreases over time. For this reason the measure provided by DGT in soils cannot be interpreted as bulk solution P concentration, as would be the case for well mixed solutions, but is a measure of P resupply from the solid soil phase (Davison et al., 2007).
Although it is usually not addressed as such, DGT essentially is a P extraction procedure. In contrast to common batch extraction techniques it can be considered a solid-state extraction that employs depletion to promote P desorption from soil. The standard DGT sampling procedure does not allow for the routine quantification of extractable P on a unit soil basis; this would require detailed and laborious consideration of P desorption kinetics, the effective diffusion coefficient in the soil, and the sampling time. Despite these methodological difficulties, DGT has the distinct advantage of providing an extractable P fraction that is defined as the fraction that can be solubilized upon depleting the soil solution. Most other batch extraction techniques lack such an unambiguous, mechanistic definition of extracted P. Furthermore, the diffusion and depletion-based sampling approach utilized by DGT mimics the diffusive uptake of P by plant roots. However, the technique falls short of mimicking processes at the root surface, which contribute to P uptake, such as P solubilization by carboxylate, and proton or phosphatase exudation.
The suitability of DGT as a soil test was demonstrated convincingly in an extensive review using literature data and mathematical simulations (Degryse et al., 2009). The authors showed that if nutrient uptake is limited by diffusion, DGT is generally a better predictor of plant uptake and nutrient deficiency than soil solution concentration or weak salt extracts. Moreover, recent 33P isotope dilution (cf. chapter 4.5) studies showed that among common agricultural soil P tests (i.e., oxalate P, Olsen P, Colwell, Bray 1, Mehlich 3, anion exchange membrane, resin P, CaCl2 P, and DGT), DGT almost exclusively measured the soil P fraction that was available for plant uptake, whereas the more aggressive extraction methods extracted large amounts of P that could not be accessed by plants (Six et al., 2012; Mason et al., 2013). Furthermore, DGT showed a much higher accuracy in predicting the P concentration in the youngest fully developed leaf in barley (Tandy et al., 2011) and in predicting yield in wheat (Mason et al., 2010) than other P tests. With little doubt, therefore, DGT is a powerful alternative to commonly used bulk soil P tests, albeit more laborious and costly. It has been calibrated for testing soils for their P status and is currently being introduced as a commercial soil P test in Australia (Australian Perry Agricultural Laboratory, Magill, Australia).
In addition to the analysis of bulk soil samples, DGT also facilitates in situ, 2D high-resolution chemical imaging of P distribution. In the 2D variant of DGT the gels are not housed in standardized sample holders but are typically placed on a plastic backing plate and covered by an ultra-thin (10 µm) membrane which serves as a very thin diffusion layer. This adaptation has the advantage of minimizing image blurring through diffusional relaxation in the diffusive layer, thereby decreasing the achievable spatial resolution below 100 µm (Lehto et al., 2012). The membrane is taped to the plastic plate and the gel assembly placed on top of a flat, water-saturated soil surface. After sampling, the resin gel is dried using a gel drier and analyzed by spatially resolved elemental analyses, e.g., laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS; Santner et al., 2012; Kreuzeder et al., 2013). By using this technique, spatial resolutions down to 50 x 50 µm2 can be achieved for sample sizes of up to 10 cm2 in reasonable measurement times of about one day. Detection limits were reported as 0.94 ng P cm−2 dried gel surface, translating to an average soil solution concentration of ≈ 3 ng P L−1 (with 24 h sampling time at 20°C).
Localized sources of P, such as microbial niches or plant roots, may be capable of resupplying P at a much higher rate than the surrounding soil (Santner et al., 2012; Ding et al., 2013) and will, therefore, be recorded in the DGT images as zones of higher DGT P concentration; this is the case even if the initial pore water concentration is the same throughout the sampled area, as confirmed by mathematical simulations (Sochaczewski et al., 2009; Santner et al., 2012). Thus, the chemical images acquired by DGT not only provide information on the spatial distribution of P, but also on changes in localized P release kinetics.
The first study using 2D DGT in soils investigated the distribution of P around the roots of two Brassica napus L. cultivars which were grown in rhizotrons (Santner et al., 2012). Plant roots and the adjacent soil were sampled with DGT for 24 h. The authors observed elevated P concentrations at the root tips and along the root axis of Brassica napus cv. Caracas (Fig. 11). The processes causing the high P concentrations at the root tips are not yet clarified. Release of P from the soil triggered by rhizosphere acidification, the release from dead root border cells or enzyme releases are potential explanations. The increased P concentrations along the root axis can be interpreted as P efflux from the roots. Furthermore, 2D DGT revealed significant differences in P concentration along roots between a P-efficient and a P-inefficient B. napus cultivar, indicating differences in their potential to deplete the soil solution of P (Santner et al., 2012).

(a) Photograph of a soil area around a root tip of B. napus cv. Caracas that was subjected to 2D diffusive gradients in thin films (DGT) analysis. The root was growing away from the soil surface into the soil, so the actual root surface is covered and not directly accessible to the DGT gel for sampling. Dotted lines give the position of the root axis, but the exact position of the root tip is unknown. (b) Corresponding 2D DGT image of the P distribution around the root with a spatial resolution of 50 x 333 µm. Values are given as CDGT P concentration in ng L−1. Substantially increased P concentrations around the tip were observed, possibly caused by an acidification of the rhizosphere and subsequent P release from the soil solid phase or by P released from dead root border cells. A few entrapped air bubbles (some indicated by arrows) caused zones of very low P concentration in the DGT image (Santner et al., 2012, unpublished). A color image is in the digital version of this article.
In summary, 2D-chemical imaging using DGT provides information on the spatial distribution and release kinetics of P in soils at the sub-mm scale. In contrast to many other chemical imaging techniques (e.g., X-ray microspectroscopy, NanoSIMS,) it does not measure soil Pt, but the labile, potentially bioavailable P fraction. This unique property, along with the capability of multi-elemental DGT analysis and the possibility to additionally map other solutes (e.g., pH, O2) using other simultaneously applied solute imaging techniques (Stahl et al., 2012; Williams et al., 2014) render DGT a powerful tool for investigating the dynamics of biogeochemical P processes in soil.
4.5 33P isotopic exchange
Isotopic approaches are powerful tools for analyzing the cycling of P between soil, plants and microorganisms. Labelling experiments can elucidate the rates of P uptake by plants (e.g., Rousk et al., 2007; Noack et al., 2014) and by microorganisms (e.g., McLaughlin et al., 1988a, b; Oberson et al., 2001; Bünemann et al., 2004; Bünemann et al., 2012; Spohn and Kuzyakov, 2013a, b). The activity of 33P in microbial biomass is usually determined the fumigation-extraction methods (cf. chapter 4.3). Recently, 33P has also been determined in phospholipids of microorganisms and plants (Rousk et al., 2007).
The short half-life of 25.3 d (33P) limits the maximum duration of labeling experiments. Nevertheless, Fardeau et al. (1996) suggested that experiments with radioactive P can extend to periods 10 times greater than the half-life; i.e., they may last up to 3 to 8 months; this is sufficient duration for measuring P availability and dynamics in soil across a vegetation season, testing the efficiency of P-fertilizers or following the decomposition of plant materials (e.g., Di et al., 1997; Wahid, 2001; Hedley and McLaughlin, 2005).
The analysis of gross Po mineralization is severely hampered by the rapid sorption of phosphate ions as well as by rapid microbial cycling of P in most soils. These problems can be overcome by utilizing 33P isotopic dilution (Frossard et al., 2011). The method is based on the addition of 33P-PO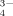 to the soil solution. The subsequent decrease in 33P activity in the soil solution is due to a dilution of the 33P - PO
to the soil solution. The subsequent decrease in 33P activity in the soil solution is due to a dilution of the 33P - PO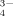 with 31P - PO
with 31P - PO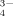 from the soil. 33P - PO
from the soil. 33P - PO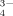 is exchanged with 31P - PO
is exchanged with 31P - PO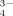 either by physicochemical processes or by biochemically driven processes such as the mineralization of Po (Oehl et al., 2001; Frossard et al., 2011). Lopez - Hernandez et al. (1998) were the first ones to separate physicochemical isotopic dilution of 33P from that due to biochemical processes; they achieved this by subtracting the former from the amount of P exchanged due to both physicochemical and biochemical processes. For this purpose, the amount of P exchanged due exclusively to physicochemical factors was modeled based on a short term (100 min) incubation experiment. The approach is based on the ssumption that the contribution of P mineralization to the isotopic dilution in the soil solution is negligible during the first 100 minutes of incubation. By dividing the amount of the biochemically mobilized P at any time t by the duration of incubation (Δt), the gross Po mineralization rate can be calculated (Oehl et al., 2001).
either by physicochemical processes or by biochemically driven processes such as the mineralization of Po (Oehl et al., 2001; Frossard et al., 2011). Lopez - Hernandez et al. (1998) were the first ones to separate physicochemical isotopic dilution of 33P from that due to biochemical processes; they achieved this by subtracting the former from the amount of P exchanged due to both physicochemical and biochemical processes. For this purpose, the amount of P exchanged due exclusively to physicochemical factors was modeled based on a short term (100 min) incubation experiment. The approach is based on the ssumption that the contribution of P mineralization to the isotopic dilution in the soil solution is negligible during the first 100 minutes of incubation. By dividing the amount of the biochemically mobilized P at any time t by the duration of incubation (Δt), the gross Po mineralization rate can be calculated (Oehl et al., 2001).
The accuracy of the calculation of the gross Po mineralization rate depends on the modelled 33P dilution. Bünemann et al. (2007) confirmed that during the first 100 min the extrapolation from the short-term experiment is valid. However, Bünemann et al. (2012) found that grassland soils with relatively high microbial activity had to be treated with HgCl2 prior to the 100 min-experiment in order to suppress the microbial activity. Other authors found that the use of HgCl2 is problematic as it alters soil physical properties (Spohn et al., 2013a). Moreover, it has been recommended to not extend the incubation time for more than 14 days in order to avoid re-mineralization of labeled Po (Frossard et al., 2011). A recent approach tries to overcome the uncertainty associated with the duration of the experiment with a numerical solution (Müller and Bünemann, 2014). Achat et al. (2010) determined gross Po mineralization by monitoring Pi and microbial P in an incubation for 500 days. While avoiding the temporal limitation, this approach is limited to soils with a very low P sorption capacity.
The gross P mineralization rate has been reported to be in the range of 0.1 to 1.9 mg P kg−1 d−1 (Oehl et al., 2001; Bünemann et al., 2007; Spohn et al., 2013a). In soils with very low concentrations of Pi in the soil solution, e.g., highly weathered tropical soils (Frossard et al., 2011), the determination of gross Po mineralization by 33P isotopic dilution is limited by the detection of Pi (approx. 0.2 mg P kg−1 soil). In these soils, in which P availability is critical for soil fertility, the development of methods to determine soluble Pi concentrations and gross Po mineralization rates is of paramount importance.
In summary, the dilution technique with radiophosphorus is a powerful tool to gain insights into P fluxes in soil. However, due to the short half-lives of 33P and 32P isotopes, radiophosphorus studies are limited in time. Furthermore, in most countries, these studies can be conducted only in the laboratory and not in the field. For field experimentation, tracing P via its stable 18O oxygen isotopes is a growing area of P research (cf. chapter 4.6).
4.6 Stable oxygen isotope ratios in phosphate extracted from soil
T (°C) = 111.4 – 4.3 × (δ18OP – δ18OW).<?h28.3>(5)
()δ18OP = fsource × δ18OP-source + (1 – fsource) × (δ18OH2O + ϵ).
()However, the generalization is not valid for acid monoesterase with fractionation factors and mechanisms being comparable to intracellular enzymes (von Sperber et al., 2014).
Our current understanding of isotopic fractionation in organisms and soil can now be used to discern the pools and processes involved in soil P cycling, especially aided by the advent of appropriate methods to separate phosphate from the matrix of environmental samples. Owing to the low solubility product of P-containing minerals, solid samples are extracted prior to a sequence of phosphate purification and separation steps. Purification procedures are required mainly because (1) O- (and P-) containing compounds in the solution might interfere with the isotope ratio of O in phosphate, and (2) the weight of the matrix that is potentially introduced in the device for isotope analysis decreases the signal-to-noise ratio.
Several preparation methods of phosphate from sediments and sea water for O isotope analysis have been published and optimized in the last two decades (Blake et al., 1997, 2005; Paytan et al., 2002; McLaughlin et al., 2004; Colman et al., 2005). In general, extracted phosphate needs to be purified through one of the following procedures (Tamburini et al., 2010): (1) ion exchange resins (Colman et al., 2005; Lécuyer et al., 2007), (2) mineral precipitations combined with resins (Kolodny et al., 1983; Shemesh et al., 1983), (3) cerium (Ce)-phosphate and resins (McLaughlin et al., 2004), or (4) Fe-oxides (Gruau et al., 2005). For soils, two approaches have been developed relying on the above-mentioned purification procedures (Zohar et al., 2010a; Tamburini et al., 2010). The method by Zohar et al. (2010a) is adapted from a procedure developed for aquatic samples (McLaughlin et al., 2004). As a first step, for solutions with low phosphate concentrations, the so-called magnesium-induced co-precipitation (MAGIC) treatment separates phosphate from the solution as a co-precipitate of Mg(OH)2 (Karl and Tien, 1992). This approach can be applied to small sample volumes without losing phosphate. In the second step, Ce-phosphate is flocculated and Ce ions are replaced by Ag ions to precipitate Ag3PO4 for δ18O analysis (Zohar et al., 2010a).
The adoption of the aforementioned purification steps to soil extracts (e.g., P fractions after Hedley et al., 1982) is hindered by the resultant strong pH changes; this alteration can facilitate the hydrolysis of Po compounds and, thus, falsify δ18OP values through the release of phosphate from the Po pool into the solution. For this reason, Tamburini et al. (2010) proposed a protocol tailored for soils that minimizes such strong pH changes and omits the MAGIC pre-concentration step. This method is based on the extraction of P from the soil with 1 M HCl followed by a combination of resins and mineral precipitation (NH4-phosphomolybdate and struvite) as purification steps. Therefore, a suitable procedure exists for the HCl-extractable and the resin-extractable P fractions in soil, where the latter is already being pre-purified by the extraction procedure itself. However, the purification for δ18OP analysis of other common soil P fractions (i.e., NaHCO3- and NaOH-extractable P) in soil remains challenging because of methodological difficulties in the complete removal of dissolved organic matter (DOM). The incomplete removal of DOM can lead to biased δ18OP values both by the presence of O-containing compounds in DOM and the hydrolysis of Po as part of DOM, which can add the isotopic signal of Po to the Pi pool (Colman, 2002; Tamburini et al., 2010). Zohar et al. (2010a) included the NaHCO3- and NaOH-extractable P fractions in their protocol, and showed that the effect of hydrolysis of Po was negligible in their samples. Because the hydrolysis of Po might depend on P speciation in soil, the use of 18O-labelled and non-labeled extraction solutions is recommended to quantify the extent of hydrolysis. However, contamination with O-containing compounds in DOM cannot be avoided completely if O/P ratios are missing. Therefore, purification procedures for soil samples with high Po concentrations are still under development.
Independent of the purification procedure, phosphate is precipitated finally as Ag3PO4 for δ18O analysis. The Ag3PO4 precipitation step was introduced by Firsching (1961) in view of its low solubility product, non-hygroscopic nature, and its rapid precipitation without the need of additional purification steps (Baxter and Jones, 1910). For the determination of the δ18OP values, Ag3PO4 is pyrolyzed at temperatures > 1,300°C in the presence of glassy carbon to produce CO which is then introduced in an isotope ratio mass spectrometer (Thermal Conversion/Elemental Analyzer-Isotope Ratio Mass Spectrometer TC/EA-IRMS). In contrast to other techniques involving fluorination of Ag3PO4 analyses, TC/EA-IRMS has the advantage that smaller sample quantities are required (about 200 to 600 μg of Ag3PO4), no dangerous chemicals (BrF5, F2 or ClF3) are needed, and the measurements are automated (Vennemann et al., 2002).
To date, not many studies on δ18OP in soil have been published which use the above-mentioned purification and measurement protocols. Fig. 12 provides an overview of published δ18OP values of various soil-relevant materials and waters. Note that this library of reference materials is still very incomplete and some of the bars in Fig. 12 represent only a few measurements (e.g., humus with only one measurement). Therefore, there is an urgent need to further extend this library with more soil-relevant materials and support ranges with more individual measurements. Angert et al. (2011; 2012) found that δ18OP values for the resin-extractable P fraction fit in the range of the expected values (δ18OP: +16 to +19.5‰) calculated from the isotopic equilibrium fractionation (Eq. 5) even if the δ18OP value of bedrock was distinctly different (δ18OP: +8 to +11‰). Similar evidence for the strong influence of intracellular enzymatic reactions on resin P δ18OP values was reported by Tamburini et al. (2012) for a soil chronosequence in a glacier forefield. In their study, even the δ18OP values of the stable P fraction (HCl-P) of the oldest site fell within the range of δ18OP values typical for reactions mediated by enzymes. The authors argued that repeated ad- and desorption processes associated with small isotopic fractionation produce this surprising finding over time (> 3,000 y), but the secondary precipitation of microbially-cycled phosphate might be an alternative explanation not mentioned in their study. In agreement with the latter speculation, Zohar et al. (2010b) highlighted biological effects on δ18OP values for the stable P fraction in an incubation experiment lasting for 31 d; also, Burmann et al. (2013) reported the involvement and potential influence of biological activity on secondary mineral formation based on δ18OP values. However, ultimate interpretations are hindered by knowledge gaps with regard to isotopic exchange between ambient water and phosphate molecules during biosynthesis in vegetation and soil organisms and their heterotrophic recycling (Tamburini et al., 2014).

Compilation of published δ18O values for various soil relevant materials and waters of different origin. Data were taken from the Tamburini et al. (2014) and references citied within. Materials marked with an asterisk were taken from Kruse and Leinweber, 2014 (unpublished).
In summary, P transformations in soil by biological activity can be traced using a stable isotope approach for O in phosphate. However, all P fractions influenced by biological activity have not yet been fully addressed in these studies because of the above mentioned difficulties in separating Po from Pi before isotope ratio measurements. Therefore, existing purification procedures must be improved or newly developed to enable δ18OP measurements also in P fractions containing high concentrations of DOM (NaHCO3-P, NaOH-P). In general, biologically-mediated processes lead to an exchange of O atoms between phosphate and ambient water. Therefore, the δ18OP of a given source (e.g., plant litter) might be equilibrated rapidly with δ18OW in soil, thereby masking the original isotopic source signal. The use of δ18OP for source identification or quantification is restricted to cases with incomplete isotopic exchange of O atoms in the phosphate molecule (Goldhammer et al., 2011). Thus, one major challenge for the successful application of δ18OP in soil science is to unravel the extent and kinetics of the exchange of O atoms between water and phosphate in soil and the underlying mechanisms, e.g., reversible vs. irreversible isotope fractionation. The combination of δ18OP with multi-isotope approaches, e.g., 32P/33P (cf. chapter 4.5), δ13Corg, δ18O-NO3/δ15N-NO3, δ18O-SO4/δ34S-SO4 might help to further disentangle the complex links between C-, N- and P-cycling in soil.
5 Synthesis and outlook
During the last decade, the continual developments and improvements in the instrumentation and capabilities of analytical methods have supported major advances in the characterization of P bonding forms, their spatial distribution and transformations in soils. These involve (1) the identification of the vast number of Po compounds, using, e.g., 2D solution-state NMR spectroscopy, and/or several high-resolution MS techniques, (2) assignments of P bonding forms and associated elements in bulk samples or samples spatially-resolved at the micro- and nanoscale resolution, using, e.g., XAS- and, XRF-spectroscopy and NanoSIMS, as well as (3) the fast screening of P concentration and bonding forms in large numbers of soil samples using vibrational spectroscopy. Novel analytical tools have been introduced for tracing soil P reactions such as (4) accessing P mobilization/immobilization reactions at the soil solution/plant/soil-interfaces using, e.g., several batch and column adsorption approaches or DGT devices, and (5) disclosing microbial P transformations by, e.g., 33P isotope dilution and δ18OP isotope ratios or by zymography.
Other advanced P methods that have not reviewed here, due to very limited applications in soil science yet, concern the following aspects of the soil P cycle: (1) small-scale genetic regulation of the soil P cycle and the corresponding identification of the P metagenome and transcriptome in soil, microorganisms and plants (e.g., Daniel, 2005; Simon and Daniel, 2011; Chhabra et al., 2013), (2) the medium-scale uptake of P by mycorrhiza and plants via, e.g., 31P magnetic resonance imaging (MRI; e.g., Pohlmeier et al., 2013; Frey et al., 2012) or 32P-autoradiogaphy (Hübel and Beck, 1993), and (3) larger scale sensing methods for the online monitoring of P fluxes in soils, soil profiles and waters, using, e.g., portable handheld XRF or IR sensors (Reidinger et al., 2012; Steffens and Buddenbaum, 2013).
Since all methods have their own specific advantages and disadvantages, the selection of a particular method or method package should be tailored to the specific objectives and limiting conditions of the study. In this regard, key factors to be considered include: (1) the speciation of P forms or soil P pools and reactions of interest, (2) the sensitivity and spatial/temporal resolution/scale, (3) the sample number/amount throughput, and, finally, (4) the cost and access to the required instrumentation and facilities. As a starting point for methods selection, Table 2 summerizes major properties, advantages and disadvantages of all methods described in this review. Furthermore, suggestions are made which part of the soil P cycle (Fig. 1) can be characterized by each method.
| Name of method | Possible applications to parts of the P cycle | Principle of measurement | Kind of results | P concentration ranges for application | Kind of sample | Sample amount required | Capable for imaging | Advantages | Disadvantages |
|---|---|---|---|---|---|---|---|---|---|
| Solid 31P NMR spectroscopy |
I1, I2, I3, P1, P3, P4, P5, R1, L1 |
NMR | total P detection | ≈ 20 µg of P / 200 mg soil sample | solid |
200 to 300 mg |
no |
– non-invasive – no extraction required – sensitive for Pi compounds |
– poor spectral resolution (large overlaps) – insensitive for Po compounds – time consuming at low P concentration |
| Liquid 31P NMR spectroscopy |
I2, I3, P1, P2, P3, P4, L1, L2, L3 |
NMR | direct P speciation | 1 µg of P per individual P species / 200 mg soil sample | solida and liquid | ≈ 200 mg | no |
– high spectral resolution – good identification of Po especially with 2D NMR |
– can be very time consuming at low P concentration – hydrolysis of Po compounds in alkaline extracts |
| Near-Infrared-spectroscopy (NIRS) |
I2, P5 |
NIR reflection/ -absorption |
total P quantification | indirect quantification by chemometric modeling, limits depending on the reference method use quantification | solid | ≈ 2000 mg | yes |
– fast – cost- and time-effective – easy to use – quantitative measurement – non-destructive |
– soil specific calibration and reference method for P concentration necessary – only indirect measurement of P |
| Mid-infrared spectroscopy (MIRS) |
I1, I2 P1, P3, P5 R1, L3 |
Vibrational or rotational energy of a molecule is enhanced upon absorption of IR radiation | mostly total P quantification, limited P speciation | ≈ 10 mg kg−1 | solid and liquid | ≈ 20 mg | yes |
– rapid screening of selected P fractions and related properties – simple sample preparation – high sample throughput (≈ 200 samples a week) |
– soil specific calibration and reference method for P concentration necessary – not working for all P fractions |
| Raman spectroscopy |
I1, I2, I3 P1, P2, P5 R1, R2, R4, L3 |
Light scattering, representing the P–O vibrations (in this context) | total P quantification, direct P speciation | ≈ 1000 mg kg−1 | solid and liquid | only a few mg | yes |
– fast – non-destructive – nearly no sample preparation – not interfered by water – detection of 16/18O isotope exchange |
– working currently for pure minerals/solutions only (disturbance by fluorescence) – sample heating by laser radiation possible |
| (Ultra-)High resolution mass spectrometry |
I2, I3 P2, P3, P4 L1, L3 |
Mass spectrometry | comprehensive P speciation, relative abundance of compounds | strongly depending on the individual method, ≈ mg kg−1 level | solida and liquid | ≈ 100 to 500 µL, soil sample amount required strongly depends on extraction method | yes (e.g., MALDI) |
– for ICR/Orbitrap: high mass resolution and mass accuracy enables sum formula assignment and resolving of complex mixtures – fast |
– destructive – no direct structural information (cannot distinguish between isomers/isobars) – only Po – suppression and discrimination of P-organics due to other compounds possible – challenging data analysis – expensive instrumentation – low sample throughput |
| (HR)-ICP-MS |
I1, I2, I3 P2, P3, P4 L1, L2, L3 |
Inductively coupled plasma coupled to mass spectrometry | total P quantification, speciation if coupled to chromatography | low mg kg−1 (in extract) | solida and liquid | ≈ 100 µL extract, soil sample amount required strongly depends on extraction method | yes (Laser-Ablation-HR-ICP-MS) |
– quantification – high throughput (if not chromatography coupled) |
– destructive – insensitive for P compared to other elements (metals) – expensive due to high gas consumption – strong matrix effects possible |
| LC-MS/MS |
I2, P1, P2, P4, R1, R3, L1, L2 |
Liquid chromategraphy coupled to tandem mass spectrometry | compound separation, qualitative information on the presence of specific compounds, compound concentrations | strongly depending on the compound under study and the extraction method prior to LC-MS/MS analysis (e.g., final glyphosate concentration in sample about 100 ng L−1) | liquid | mostly depending on the compound concentration in sample, typical injection volumes range from 10 to 50 µl | no |
– highly selective and sensitive in the Selected Reaction Monitoring (SRM) mode – enables highly quantitative and reproducible results |
– destructive detection method – expensive and high maintenance instrument – limited structural information on the compounds in the SRM mode |
| Direct Infusion Nanospray Quadrupole Time-of-Flight MS (Q-TOF MS/MS) |
I2, P2, P3, P4, R2, R3, L2, L3 |
Mass spectrometry | comprehensive speciation and quantification of P lipids | 0.25 nmol g−1 | solida and liquid | ≈ 1000 to ≈ 5000 mg | no |
– direct quantitative measurement of phospholipid molecular species in their native structure – fast and simple extraction procedure – high sample throughput – very low detection limit |
– no information about the position of double bonds in fatty acids – expensive instrumentation |
| Nano-scale secondary ion mass spectrometry (NanoSIMS) |
I1, I2, I3 P1, P2, P3, P4, P5, L1, L3 |
Secondary ion mass spectrometry | relative abundance without speciation | ≈ mg kg−1 or better is possible | solid | a few mg | yes (spatial resolution: up to 50 nm) |
– parallel detection of up to 7 masses enables co-localization – stable isotopes abundances (e.g., 13C, 18O) can be analyzed simultaneously |
– only vacuum stable sample – sufficient sample preparation is essential – quantification/speciation analysis not yet established – up-scaling of results can be challenging – restricted access to facilities |
| Fluorescence (XRF) spectroscopy |
I1, I2, I3 P2, P5, L3 |
X-Ray fluorescence | P quantification | ≈ 10 mg kg−1 | solid and liquid | a few mg | yes (spatial resolution: up to 50 nm) |
– fast – non-destructive – simultaneous multi-element detection – high sample throughput even in the field |
– quantitative, but no qualitative P information – restricted access to beamlines (synchrotron-based XRF) |
| X-ray absorption spectroscopy (XAS) |
I1, I2, I3 P1, P2, P5, L1, L2, L3 |
X-Ray absorption | direct P speciation and quantification | > 5000 mg kg−1 (L2,3-edge); > 100 mg kg−1 (K-edge) | solid and liquid | a few mg | yes (spatial resolution: up to 50 nm) |
– non-destructive – no additional sample preparation – element specific qualitative and quantitate detection |
– low sensitivity in soils – high detection limit, especially P L2,3-edge – fit results sometimes ambiguously – restricted access to beamlines – low sample throughput |
| X-ray photoelectron spectroscopy (XPS) |
I2, I3 P1, P5, L3 |
X-Ray absorption | direct P speciation and quantification | similar to XAS, depending on concentration of P on the sample surface | solid and liquid | a few mg | yes (spatial resolution: up to 50 nm) |
– non-destructive – no additional sample preparation – element specific qualitative and quantitate detection – high surface sensitive ≈ 10 to 15 nm depth |
– only surface sensitive – no bulk information – limited P speciation due to overlaps of chemical shift |
| Sorption isotherms |
P1, R1, R4 |
Equilibrium reactions; photometric/ICP measurements | operationally defined fractions | depending on the method use for P quantification | solid | solid:solution ratio 1:2 to 1:4 (w:v) ≈ 500 mg (batch) : ≈ 2000 mg (continuous flow) | no |
– low cost easy to apply (batch & continuous flow) – better mimicking soil conditions such as higher soil/solution ratio (continuous flow) – dynamic flow removal of reaction products (continuous flow) |
– very time and labor intensive (batch and continuous flow) – results highly effected by experimental conditions such as contact time, temperature and initial concentration (batch & continuous flow) – no removal of reaction products (batch) |
| Quantum-chemical modeling |
P1, P2, R1, R4 |
Quantum-chemical calculations | models showing fundamental physical properties, quantitative structure-activity-relationships | all phases | no |
– atomistic insight – explanation of experimental results, e.g., of sorption isotherms |
– systems often not well-defined – complicated soil solid phase difficult to reflect in current models |
||
| Microbial P |
P4, R3 |
Photometric/ICP measurements | microbial P concentration | depending on the photometric method used for quantification | solid and liquid | ≈ 50 g | no |
– low cost easy to apply – suitable indicator of this labile P fraction |
– low sensitivity – not suitable for soils with high P sorption |
| Colorimetric phosphatase assays | R2, R3 | Photometric | product concentration after use of artificial substrates in defined conditions | depending on the photometric method used for quantification | solid and liquid | ≈ 10 g | no |
– low cost – easy to apply – suitable indicator for potential P mobilization activity from organic matter |
– only indication of a potential activity under defined environmental conditions – in situ affected by diverse environmental properties |
| Soil zymography of phosphatases | R2, R3 | Hydrolysis of fluorescent substrate | Distribution of phosphatase activity in soil | solid | some g | yes (spatial resolution: 1–2 mm) |
– allows for measuring the distribution of phosphatase activity in-situ – non-destructive imaging method |
– requires a smooth soil surface | |
| Diffusive gradients in thin films (DGT), bulk soil analysis | P1, P2 | Photometric/ICP measurement | operationally defined fractions | depending on the method used for P quantification | solid and liquid | a few g | no |
– measures the labile (reversibly adsorbed) soil P fraction – highly correlates to plant uptake |
– time-consuming – cannot mimic root exudation processes |
| Diffusive gradients in thin films (DGT), 2D imaging using LA-ICP-MS | P1, P2 | Laser ablation ICP-MS | operationally defined fractions | 0.94 ng P cm−2 dried gel surface, equal to ≈ 3 ng P L−1 in soil solution (24 h sampling time, 20°C) | solid | flat soil surface (20+ cm2) | yes (spatial resolution: 50x50 µm) |
– allows for measuring the spatial distribution of labile (reversibly adsorbed) soil P, e.g., around plant roots – mapping of additional parameters (inorganic anions and cations, pH, O2, etc.) is possible |
– time-consuming – expensive instrumentation – no differentiation between Po and Pi – cannot mimic root exudation processes |
| 33P isotopic exchange |
L3, R1, R2 |
Isotopic exchange | gross Po mineralization rate | depending on the method used for PO4 quantification | solid | a few g | no | – practically the only way to determine gross Po mineralization in a strongly P-sorbing soil | – gross P mineralization can be measured only over a few weeks |
| Stable oxygen isotope ratios |
I3, P2, P4, P5, L2 R2, R3 |
Mass spectrometry | operationally defined fractions | 0.02 to 0.6‰ | solid and liquidb | strongly depending on extraction method, after extraction 2.5 µmol PO4 equal to ≈ 200 µg Ag3PO4 | yes (cf. NanoSIMS) | – transformation processes of P can be identified on a virtually molecular basis |
– purification procedure labor- and time-consuming – Pi separation difficult in samples containing Po |
- aafter extraction
- bafter extraction and precipitation as Ag3PO4
While perhaps sounding clichéd, it should be emphasized once again that applying just a single method often will be insufficient to comprehensively characterize the broad range of P forms and pools in soil. Therefore, multi-methodological approaches are essential to integrate and enhance the individual advantages of specific methods (e.g., Negassa et al., 2010; Giguet-Covex et al., 2013; Hashimoto et al., 2014; Liu et al., 2013). Essentially, in addition to the sophisticated molecular-level speciation, or spatially-resolved techniques reviewed here, such multi-methodological approaches should involve complementary, rapid and inexpensive traditional wet chemical methods, e.g., selected agronomic and environmental soil P tests, sequential fractionations (Negassa and Leinweber, 2009) and/or alternative routine sensing methods (e.g., IR).
Adopting a more holistic approach to soil P characterization may also forestall researchers from instinctively applying purely technique-based innovations that run the risk of diverting attention from the central issue: the actual and foreseen global impacts of shortages in plant available P, viz food security, the bio-economy and freshwater quality.
Acknowledgements
The authors are thankful to the German Soil Science Society (DBG) for the financial support of the workshop “Innovative Methods in Soil Phosphorus Research” held at Rostock. The workshop and the research of some co-authors have been coordinated in the frame of the Leibniz ScienceCampus “Phosphorus Research” in Rostock (http://www.wissenschaftscampus-rostock.de). The authors are thankful to Isabell Gauthier, Dr. D. Abdala, and Pee Narf for providing some of the XAS spectra of reference standards and again Isabell Gauthier for very helpful comments and suggestions to improve the manuscript. X-ray absorption spectra shown in this article were recorded at the Canadian Light Source (PGM and SXRMB beamlines), which is funded by the Canada Foundation for Innovation, the Natural Sciences and Engineering Research Council of Canada, the National Research Council Canada, the Canadian Institutes of Health Research, the Government of Saskatchewan, Western Economic Diversification Canada, and the University of Saskatchewan, and the SXS beamline at the Brazilian Synchrotron Light Laboratory (LNLS); beamline scientist in charge provided helpful comments on the manuscript. The NanoSIMS at the Leibniz Institute for Baltic Sea Research (IOW) was funded by the German Federal Ministry of Education and Research (BMBF), grant identifier 03F0626A. The work by Dr. Jakob Santner was funded by the Austrian Science Fund (FWF): P23798-B16. We are grateful for the disposal of the bacterial samples of N2-fixing bacteria by Dr. habil. Katarzyna Hrynkiewicz (Nicolaus Copernicus University, Torun, Poland) financially supported by a grant from the National Science Centre (Poland) (2012/07/B/NZ9/01801). Funding of the project (FOR1320, coordinator U. Köpke) by the German Research Foundation (DFG) is gratefully acknowledged. The data for NIRS model calibration, shown in this article, was provided by Björn Todt (Chair of Silviculture, Albert Ludwig University Freiburg, Germany) who was supported by a DFG grant (BEF China, FOR 891). Project funding of Jörg Niederberger was provided by the Thünen Institute, Eberswalde. The δ18O data from Kruse and Leinweber (unpublished, 2014) are based on a project funded by the FNR (FNR 22400112). Finally, the authos are very grateful to three anonymous reviewers for helpful comments to improve the manuscript.

