Dementia associated with disorders of the basal ganglia
Funding information
KSV is funded by a Medical Research Council (MRC) UK studentship (MR/N013867/1); KMS is funded by an Alzheimer Fonden, Sweden studentship; DPB is funded by a Consejo Nacional de Ciencia y Tecnologia (CONACyT), Mexico studentship; JB is funded by a Biotechnology and Biological Sciences Research Council (BBSRC) UK studentship; and DMC is funded by the Dementia Research Institute (UCL).
Abstract
Dementia is now the leading cause of death in the United Kingdom, accounting for over 12% of all deaths and is the fifth most common cause of death worldwide. As treatments for heart disease and cancers improve and the population ages, the number of sufferers will only increase, with the chance of developing dementia doubling every 5 years after the age of 65. Finding an effective treatment is ever more critical to avert this pandemic health (and economic) crisis. To date, most dementia-related research has focused on the cortex and the hippocampus; however, with dementia becoming more fully recognized as aspects of diseases historically categorized as motor disorders (e.g., Parkinson's and Huntington's diseases), the role of the basal ganglia in dementia is coming to the fore. Conversely, it is highly likely that neuronal pathways in these structures traditionally considered as spared in Alzheimer's disease are also affected, particularly in later stages of the disease. In this review, we examine some of the limited evidence linking the basal ganglia to dementia.
Significance
Dementia now accounts for over 12% of deaths in the United Kingdom, exceeding those caused by cancer or heart disease. With the risk of developing dementia doubling every 5 years from the age of 65 and a globally aging population, finding a cure or an effective treatment to delay onset or progression is ever more imperative. Basal ganglia structures have historically been overlooked but it is becoming increasingly apparent that they demonstrate both dysfunction and subsequent neurodegeneration in various dementias. We summarize some of this currently sparse evidence as a means to highlight areas for further research.
1 INTRODUCTION
The basal ganglia are a series of interconnected subcortical structures in the forebrain consisting of several components, including the striatum (comprised of the caudate nucleus and the putamen), the globus pallidus (internal [GPi] and external [GPe] segments), the subthalamic nucleus, and the substantia nigra (pars compacta [SNc] and pars reticulata [SNr]) (Graybiel, 2000). The primary roles of the basal ganglia are motor control and motor learning; however, emotional processing and goal-directed motivation have also been related to basal ganglia functions, indicating the multifactorial role of the basal ganglia in a number of different behaviors. Given their central role in motor control, impairments in the basal ganglia have been associated with a number of neurodegenerative disorders which display altered motor behavior, including Parkinson's disease (PD), dementia with Lewy bodies (DLB) and Huntington's (HD) and Alzheimer's diseases (AD). In particular, degeneration of striatal neurons has been associated with HD, whereas loss of neurons in the substantia nigra has been related to PD and similar disorders (Bernheimer, Birkmayer, Hornykiewicz, Jellinger, & Seitelberger, 1973). In this review, we will first briefly describe the basal ganglia circuit, followed by a discussion of how impairments in basal ganglia structures can contribute toward disease development.
2 BASAL GANGLIA CIRCUIT
Much research has focused on elucidating the mechanism through which the basal ganglia structures interact and has provided a much better understanding as to how their activity and organization may be affected under neurodegenerative conditions. The basal ganglia receive their main inputs through the striatum and subthalamic nucleus. These structures receive afferents from the cortex, thalamus, and brainstem regions, placing the basal ganglia in a central position for integrating motor information and mediating the functions of the forebrain (Graybiel, 2000).
2.1 Input nuclei
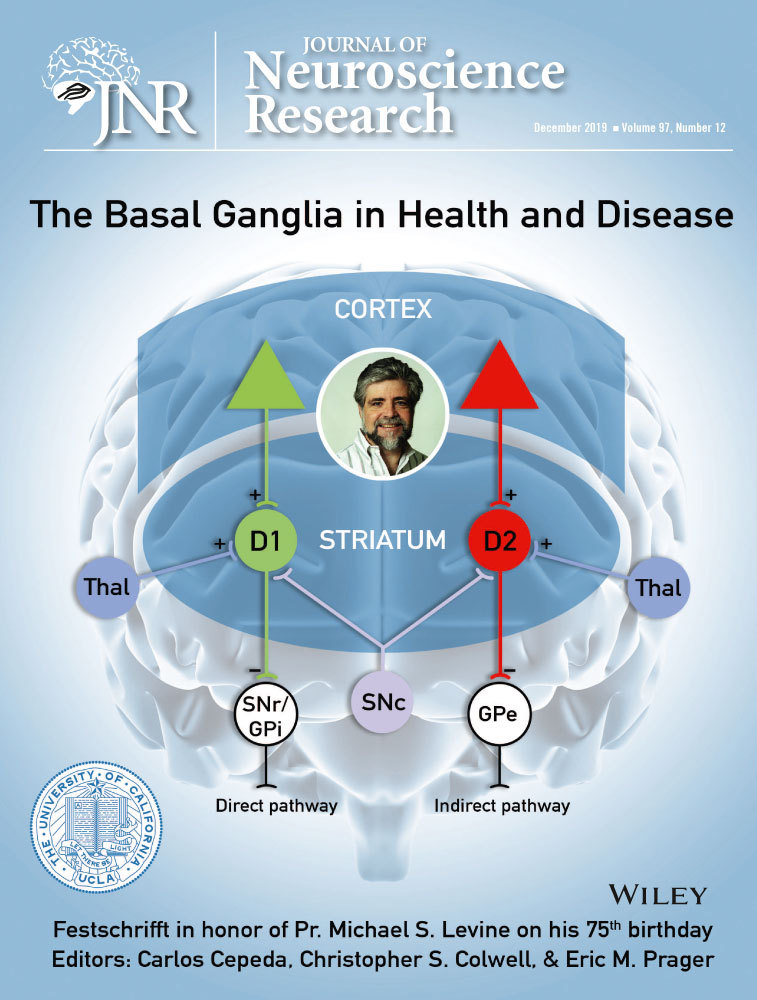
The striatum, the main input nucleus of the basal ganglia, is primarily made up of ϒ-aminobutyric acid (GABA)-ergic medium-sized spiny neurons (MSNs), neurons with high abundance of dendritic spines comprising around 90% of all striatal neurons and a small proportion of cholinergic and GABAergic interneurons (Kawaguchi, 1993). These neurons relay the excitatory input they receive from cortical regions either monosynaptically to the output region of the basal ganglia, that is the SNr or GPi, or to the GPe which subsequently projects to the output structures. The classical view is that the striatal neurons directly innervating the output regions of the basal ganglia express dopamine receptor subtype 1 (D1) receptors and form the direct pathway. On the other hand, the latter neurons, which project to the globus pallidus, contain dopamine receptor subtype 2 (D2) receptors and make up the indirect pathway (Albin, Young, & Penney, 1995; DeLong, 1990). This expression pattern allows for the dopaminergic neurons in the SNc to modulate striatal neuron activity. Dopamine release increases the activity of the direct pathway through the activation of D1 receptors, while also decreasing the activity of the indirect pathway through D2 receptor activation through L-type calcium channels (Hernandez-Lopez, Bargas, Surmeier, Reyes, & Galarraga, 1997; Kravitz et al., 2010). Ultimately, despite the seemingly opposing actions of these pathways, the net outcome of dopamine release results in the activation of cortical motor regions.
The subthalamic nucleus, composed predominantly of glutamatergic neurons, mainly receives corticothalamic inputs, as well as projections from within the basal ganglia and specifically, from the GPe. This region projects to the globus pallidus or SNr, a projection known as the hyperdirect pathway, which has been suggested to be involved in the regulation and cessation of motor behavior and cognition (Nelson & Kreitzer, 2014; Wessel & Aron, 2013; Wessel, Conner, Aron, & Tandon, 2013; Wessel et al., 2016). Interestingly, the subthalamic nucleus has been highlighted as a target for deep brain stimulation in PD patients due to its ability to directly relay motor and premotor cortical information to the globus pallidus through this hyperdirect pathway (Dayal, Limousin, & Foltynie, 2017; Hamel et al., 2003).
2.2 Output nuclei
The two main output regions of the basal ganglia are the GPi and the SNr. They are primarily composed of GABAergic neurons and therefore form inhibitory projections with several thalamic and brainstem nuclei. The GPi and SNr are innervated by inhibitory projections coming from striatal neurons through the direct pathway, as well as excitatory projections from the subthalamic nucleus through the indirect pathway (Deschenes, Bourassa, Doan, & Parent, 1996). Some of the projections of the output nuclei are directed at the intralaminar nuclei of the thalamus which comprises both motor and limbic circuits, highlighting the potential involvement of the basal ganglia in cognitive functions as well as motor control (Fazl & Fleisher, 2018).
The tight organization of the basal ganglia demonstrates the high interconnectivity of their circuits and how impairment of one region may lead to abnormal functioning of another. In fact, exactly this interdependency of basal ganglia activity is what has been shown to be a driving factor for several neurodegenerative diseases, as discussed below.
3 THE BASAL GANGLIA IN NEURODEGENERATION AND DEMENTIA
The basal ganglia consist of a range of subcortical cell groups that serve together to drive a variety of functions, including motor control and learning, working and procedural memory, executive functions, such as attentional filtering and emotions (Lanciego, Luquin, & Obeso, 2012). As previously outlined, due to the diverse range of inputs and outputs to and from these deep embedded nuclei, damage to these circuits may affect any number of functions. Disorders of the basal ganglia have helped with the understanding of their role, as many of them share a common pathological profile and have an overlap of cognitive and motor symptoms. The basal ganglia are particularly susceptible to neurodegeneration, evidenced by the fact that many neurodegenerative disorders have, to an extent, some form of damage to basal ganglia circuitry. For example, PD is characterized by the progressive loss of dopaminergic neurons in the substantia nigra and basal ganglia network, causing loss of control of voluntary movement (Poewe et al., 2017), whereas degeneration of MSNs in the striatum is the predominant basal ganglia pathology in HD (Ehrlich, 2012). Corticobasal degeneration is a rare progressive disease involving neuron loss and atrophy of multiple areas of the brain, including the cerebral cortex and the basal ganglia. Patients with corticobasal degeneration have overlapping symptoms with multiple other dementias, most notably progressive supranuclear palsy but also PD, AD, and DLB (Reich & Grill, 2009). Improving knowledge of any shared pathology underlying these dementias will no doubt aid the search for effective therapies.
3.1 A common pathway in the basal ganglia underlying psychiatric disorders and dementia
Dementia refers to a set of symptoms caused by brain disease or injury, resulting in persistent memory, personality, mood, and judgment impairment. Many of the neurodegenerative diseases, including AD, PD, HD, and DLB, are characterized by deterioration into some form of dementia sooner or later over the course of the disease. Cell death caused by abnormal protein accumulation and aggregation is a common feature of these diseases, which then leads to cognitive disturbances and decline into dementia.
The basal ganglia interact closely with the frontal cortex and have crucial roles in cognition and behaviors such as judgment and reasoning (Frank, Loughry, & O’Reilly, 2001) and damage to the basal ganglia can lead to some of the same cognitive impairments as damage to the frontal cortex. This is evidenced in individuals with frontotemporal dementia, particularly frontotemporal lobar degeneration, where atrophy in the frontal and temporal lobes of the brain leads to a profound decline in behavior and language ability (Rabinovici & Miller, 2010). Both frontotemporal dementia and frontotemporal lobar degeneration appear to have a shared pathological profile with certain psychiatric disorders, particularly bipolar disorder and major depressive disorder (Nascimento et al., 2019). This indicates that there may be a common underlying pathway driving cognitive disturbances in dementia and psychiatric disorders; the basal ganglia are highly likely to be involved as they have been shown to have abnormalities in both. For example, significant shape differences are observed in the striatum in bipolar disorder (Hwang et al., 2006) and frontotemporal dementia patients exhibit structural changes in the striatum and thalamus (Bede et al., 2018).
Another psychiatric disorder that dementia may have overlapping mechanisms with is depression. Individuals with disorders of the basal ganglia are particularly susceptible to depression and patients with AD, PD, HD, DLB, frontotemporal lobar degeneration, and vascular dementia, to name a few, commonly experience depressive symptoms at some point during the disease (Baquero & Martin, 2015; Lanctot et al., 2017; Ring & Serra-Mestres, 2002). Up to 50% of PD (Cummings, 1992) and 30% of HD patients (Slaughter, Martens, & Slaughter, 2001) have depression, making it likely that basal ganglia damage in neurodegeneration may impact pathways underlying depression. Depression may also be a risk factor for dementia (Stern, Marder, Tang, & Mayeux, 1993) and neurodegenerative abnormalities in the basal ganglia may contribute to depression: in patients with major depressive disorder, length of illness, and number of depressive episodes is associated with decreased left putamen and left globus pallidus gray matter volume (Lacerda et al., 2003). This suggests that basal ganglia damage impacts higher level cognitive functions that are affected in both depression and dementia. Understanding this link will help elucidate if drugs used to treat depression can alleviate some of the symptoms of dementia and vice versa.
3.2 Basal ganglia and cognitive function in dementia
Individuals with dementia have a wide range of cognitive impairments, including memory loss as one of the first signs. This may be linked to the basal ganglia in some way due to their role in higher level cognitive functions. Procedural memory, or the ability to learn new skills, is particularly affected in PD and HD patients. In early PD, spatial learning and working memory, typically associated with nigrostriatal and dopaminergic pathways, are selectively impaired, indicating damage to networks connecting the prefrontal cortex and basal ganglia (Postle, Locascio, Corkin, & Growdon, 1997). In a study of PD patients with or without dementia, dementia was linked to an increased atrophy of regions of the basal ganglia including the caudate and the putamen (Melzer et al., 2012). Overall this suggests that damage to the basal ganglia may drive certain symptoms of dementia but how this occurs remains unclear.
In the healthy brain, the basal ganglia may regulate the ability to multitask and filter out distractions: individuals with better working memory have increased activation in the globus pallidus when faced with distracting stimuli, suggesting an improved efficiency at filtering out unwanted information (McNab & Klingberg, 2008). AD patients in particular have impaired working memory and attentional deficits (MacPherson, Della Sala, Logie, & Wilcock, 2007). It is therefore conceivable that basal ganglia pathways controlling executive function and memory are affected in neurodegenerative disorders.
In a drive to develop novel, effective, and targeted therapies for the dementias which currently have limited to no cure, it is crucial to understand how specific brain areas are impacted and how this may be shared between diseases. This review addresses shared basal ganglia dysfunction across neurodegenerative diseases and dementias and their implications.
4 A NOTE ON GENETIC RISK FACTORS OF DEMENTIAS
It is becoming ever clearer that a whole host of genetic variants contribute to one's risk of developing dementia. A huge driver in the understanding of genetic risk factors has been the now relative ease and low cost of performing genome-wide association studies (GWAS). These studies are gaining more statistical power as meta-analyses are performed, combining studies to create large data sets. It has become clear that there is limited overlap in the risk genes for the various diseases (Brainstorm Consortium et al., 2018) and this may contribute to the regional specificity of neurodegeneration associated with each disease. While we do not intend to offer an exhaustive review of the genetics that have been associated with dementias related to basal ganglia diseases, we discuss some of the key risk factors for each disease and refer the reader to recent reviews for a more extensive discussion.
5 PARKINSON’S DISEASE
Among the dementias in which the basal ganglia are affected, PD has been the most extensively studied. PD occurs in between 5 and 35 in 100,000 individuals worldwide, with incidence increasing after the age of 65 (Poewe et al., 2017). It is a neurodegenerative disease which is associated with the progressive loss of midbrain dopaminergic neurons, coupled to slowness of motor movements (bradykinesia), rigidity, tremor, as well as postural defects. In addition to these motor defects, PD patients also demonstrate a number of non-motor symptoms, including depression, anxiety, and in most cases, dementia (Hely, Reid, Adena, Halliday, & Morris, 2008). The earliest symptoms of PD, both motor and non-motor, progress slowly and are therefore very difficult to detect. The accumulation of protein aggregates, known as Lewy bodies, within and around neurons is a common pathological hallmark (Spillantini et al., 1997). Examinations of postmortem brains of patients have demonstrated that these accumulations contain misfolded and aggregated α-synuclein as well as other proteins, including hyperphosphorylated tau and amyloid-β in cases where patients are diagnosed with DLB (Wills et al., 2010). Perhaps the most prominent and unifying morphological feature of PD is the extensive loss of dopaminergic neurons in the SNc and hence their dopaminergic projections to the striatum (Hughes, Daniel, Ben-Shlomo, & Lees, 2002). Given that these projections have been highlighted as important modulators of striatal and hence basal ganglia activity and output to the motor cortex, their impairment has been postulated to play a key role in the emergence of the motor defects associated with PD.
5.1 Genes associated with Parkinson’s disease
PD itself is a multifactorial disease, arising from both genetic and environmental factors, which together contribute to disease progression through their effects on the accumulation and aggregation of misfolded proteins, clearance mechanisms, mitochondrial damage, and neuroinflammation. However, although more than 90% of cases are sporadic, a proportion of patients have a family history of the disease. To date, a number of genes have been identified as causative for PD, including mutations in the genes encoding for PARKIN, PINK1, SNCA (α-synuclein), DJ-1, and LRRK2. These proteins have been shown to play an important role in molecular pathways, which, when perturbed, can lead to neurodegeneration (Schapira, 2008). For instance, PINK1, DJ-1, and LRRK2 have been shown to encode proteins localized to mitochondria, suggesting their potential involvement in mitochondrial function and dynamics (Hao, Giasson, & Bonini, 2010; Pickrell & Youle, 2015; Truban, Hou, Caulfield, Fiesel, & Springer, 2017). We refer the reader to the review by Karimi-Moghadam, Charsouei, Bell, and Jabalameli (2018). Animal models of PD have contributed much to the current understanding of how mutations in these proteins can lead to mitochondrial impairment, oxidative stress, and disturbance in calcium homeostasis, which together can bring about disease pathology (Klein & Westenberger, 2012; Lill, 2016; Polymeropoulos et al., 1997).
5.2 Loss of dopamine and physiological changes in the basal ganglia in Parkinson's disease
Loss of nigrostriatal dopaminergic neurons is a prominent feature of the disease. Interestingly, the dopaminergic neurons projecting to the striatum are more largely affected than those projecting to limbic regions, explaining the disturbances seen primarily in motor control (Galvan, Devergnas, & Wichmann, 2015; Kish, Shannak, & Hornykiewicz, 1988). This loss of dopaminergic neurons occurs in the early stages of the disease, with indications of this phenotype to precede the diagnosed onset of motor symptoms, suggesting their crucial role in modulating motor behavior (Iacono et al., 2015; Poewe et al., 2017). A common diagnostic tool for PD is the use of the radiolabeled ligand 123I-ioflupane in single-photon emission tomography, which binds to dopamine transporters in the presynaptic space. The 123I-ioflupane signal is reduced in the SNc and striatum of PD patients, which correlates well with the disease state, highlighting the importance of dopaminergic neuron death in driving pathology (Brooks, 2016; Kraemmer et al., 2014; Marshall et al., 2009).
The specific susceptibility of these neurons has been suggested to be due to a number of factors, particularly their high energetic demand, rendering them highly susceptible to mitochondrial dysfunction and oxidative stress, as well as their high dependence on L-type Ca2+ channels for their activity, making them vulnerable to altered Ca2+ influx (Bishop et al., 2010; Chan et al., 2007; Foehring, Zhang, Lee, & Callaway, 2009; Liss et al., 2005; Sulzer, 2007). The importance of these neurons is highlighted by the efficiency of L-DOPA, a dopamine precursor, at slowing, albeit not halting, disease progression (Barbeau, 1969). Moreover, the finding that nigrostriatal dopaminergic neurons are also selectively vulnerable to MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine), a toxin which leads to the development of a movement disorder very similar to PD, further highlights the pivotal role these neurons play in PD pathogenesis (Varastet, Riche, Maziere, & Hantraye, 1994).
SNc dopaminergic neurons harbor extensive axonal arbors that contact a large number of synapses, making their axons highly susceptible to alterations in axonal trafficking and abnormal mitochondrial activity (Arbuthnott & Wickens, 2007). Thus, due to the high metabolic demand of axonal terminals, somatodendritic regions of these neurons become less populated with mitochondria (Liang, Wang, Luby-Phelps, & German, 2007), which reduces their ATP-generating capacity and may subsequently result in an energy crisis (Nicholls, 2008). In support of this idea, human imaging and postmortem studies have demonstrated that some changes in the basal ganglia, such as the emergence of dysfunctional nigrostriatal axonal terminals (Kordower et al., 2013) and changes in dopamine turnover (Sossi et al., 2004), occur even before disease diagnosis. Recent positron emission tomography imaging studies have also demonstrated the loss of vesicular monoamine transporter 2 activity in the posterior putamen of patients with mild PD (Fazio et al., 2015; Hsiao et al., 2014). These findings suggest a reduced innervation of striatal neurons by SNc dopaminergic neurons and hence support the idea of an early loss of their axonal terminals. Furthermore, α-synuclein, the main component of the pathological Lewy bodies, has also been suggested to play a role in the early abnormal axonal pathology observed in PD patients through a yet unknown mechanism. This idea has arisen from the protein's increased propensity for fibrillation when mutated (Conway, Harper, & Lansbury, 1998; Narhi et al., 1999), its ability to regulate dopamine metabolism and transmission (Chen et al., 2008; Lam et al., 2011; Perez et al., 2002), and its toxic influence on mitochondrial function, as shown in animal models (Martin et al., 2006; Ordonez, Lee, & Feany, 2018). Given that α-synuclein is in fact a synaptic protein and is therefore able to exert its toxic effects at synaptic sites, it is reasonable to suggest its potential role in dopaminergic axonal dysfunction in the disease.
The dopaminergic neurons in the SNc are autonomously active, generating broad action potentials (2–10 Hz) in the absence of synaptic input (Chan et al., 2007; Grace & Bunney, 1984; Mamelak, 2018). In the basal ganglia, this pacemaker activity is considered important for the maintenance of basal dopamine levels and is highly reliant on the activity of voltage-dependent L-type Ca2+ channels with the Cav1.3 subunit (Striessnig et al., 2006). This subunit allows for Ca2+ channels to be open during a hyperpolarized state, resulting in continuous oscillations of intracellular Ca2+ levels. However, this continuous Ca2+ influx can overload the Ca2+ buffering ability of SNc dopaminergic neurons and thus may be considered a key contributor to their susceptibility in PD (Surmeier, Guzman, Sanchez, & Schumacker, 2012). Ca2+ influx requires an equivalent amount of ATP molecules for its exclusion, making dopaminergic neurons at a constant high energetic demand. In fact, nigrostriatal dopaminergic neurons have also been shown to have a low Ca2+ buffering capacity resulting from their relatively low levels of Ca2+-binding proteins such as calbindin. This allows excess intracellular Ca2+ to bind to intracellular proteins, which can trigger cell death pathways and further enter the mitochondria to then exert continuous mitochondrial oxidant stress (German, Manaye, Sonsalla, & Brooks, 1992; Surmeier et al., 2017). In comparison, the dopaminergic neurons in the ventral tegmental area, which are relatively resistant to pathology, express high levels of this protein, highlighting the importance of Ca2+ buffering capacity in cell susceptibility in PD.
It is important, however, to highlight that all the above mechanisms happen simultaneously, making it particularly challenging to target just one mechanism to improve neuronal viability. The fact that current treatments have been ineffective stresses the importance to understand what makes the basal ganglia susceptible in this disease and how they can be targeted for symptom alleviation.
6 HUNTINGTON’S DISEASE
HD is characterized by chorea, involuntary and continuous movements of the body, especially of the extremities and face. Clinically, it consists of choreoathetosis, dementia, and behavioral syndromes. It is an autosomal dominant progressive neurodegenerative disorder caused by impairments in the basal ganglia and cortex. In the basal ganglia, there is a selective degeneration of MSNs, resulting in the lack of inhibition of undesired movements and leading to cognitive, behavioral, and motor decline (Reiner, Dragatsis, & Dietrich, 2011).
HD onset is around the age of 40 and early symptoms include depression, small uncontrolled movements, poor coordination, learning difficulties, and behavioral changes. As the disease progresses, the undesired movements become more pronounced and daily activities like walking, speaking, or swallowing are affected. Additionally, HD can also be found in the younger population (<20 years) with similar symptoms but with some variations, for instance hypokinetic signs, rigidity, drooling, or seizures (Roos, 2010; Tereshchenko et al., 2019).
The genetic cause for this disease is clear: mutations in the HTT gene located on chromosome 4, which is responsible for the production of the huntingtin protein, involving the DNA segment CAG trinucleotide repeat in exon 1. Affected individuals possess a sequence of more than 36 CAG repeats and longer expansions result in earlier disease onset. The CAG codon encodes glutamine and thus results in longer versions of huntingtin. In turn, this abnormal protein is cleaved into toxic fragments that disrupt normal neuronal functions and eventual death of projection neurons of the basal ganglia occurs (Reiner et al., 2011). More recently, it has become apparent through GWAS that disease presentation is moderated through variants of genes other than HTT. A review is provided in Holmans, Massey, and Jones (2017).
6.1 Structural alterations of basal ganglia in Huntington's disease
Brain abnormalities in HD develop before evident symptoms or clinical diagnosis. There is 25% brain weight loss at advanced stages of the disease. The neuropathology consists of atrophy, neuronal loss, and astrogliosis of the basal ganglia and cortical structures. Striatal pathology starts caudally and then spreads rostrally in a dorsomedial to ventromedial direction (Fazio et al., 2015). Additionally, other regions of the brain are affected while the disease progresses, such as hippocampus, amygdala, and cerebellum (Rosas et al., 2003).
The loss of striatal and pallidal neurons in HD is selective; early in the disease there is loss of putaminal GABAergic cells projecting to the lateral globus pallidus, which affects the indirect pathway of the basal ganglia. The balance between indirect and direct pathways is consequently in favor of the direct pathway: there is unsuccessful inhibition and thus excitation of thalamic neurons is enhanced, resulting in random and inappropriate neuronal activity. In contrast, when the disease is advanced, cholinergic signals are lost and the caudate nucleus is atrophied. Detection of this state of the disease is visible through MRI or computerized tomography in the brain, where enlarged lateral ventricles of the caudate frontal horns are observed (Reiner et al., 2011).
In HD, there is also a significant shrinkage of the amygdala. This region plays a crucial role in the limbic circuit of the basal ganglia to trigger specific cognitive responses linked to motor activities. Indeed, declining emotion recognition in patients correlating with amygdala volume loss has been reported (Kipps, Duggins, McCusker, & Calder, 2007). On the other hand, the thalamus, subthalamus, and substantia nigra undergo cell loss, shrinkage, and astrogliosis as well but at lower levels. About 40% of GABAergic and dopaminergic neurons of the substantia nigra are lost, which could contribute to the akinesia symptoms seen in both HD and PD (Vonsattel, 2008).
7 ALZHEIMER’S DISEASE
It is estimated that there are currently over 50 million people in the world living with dementia, with the number expected to triple to 152 million by 2050 (Patterson, 2018). AD is the most common cause of dementia, accounting for over 60% of all dementia cases. The global and societal impact of dementia is huge and there are still no effective treatments in halting or improving symptoms of AD, so understanding the pathology of this disorder represents a crucial step toward finding a cure.
Over 97% of AD cases are sporadic and occur later than 60 years of age. No common causative gene has been identified for the late onset disease; however, the APOE gene influences risk of developing the disease, with the ε4 allele increasing and the ε2 allele decreasing risk with respect to the ε3 allele (Verghese, Castellano, & Holtzman, 2011). However, sporadic AD is thought to arise from a complex interplay between genetic and environmental factors. There are three genes in which mutations have been identified that cause rare familial forms of AD: amyloid precursor protein (APP), presenilin 1, and presenilin 2 (PSEN1/PSEN2). More recently, GWAS have highlighted a number of risk factor variants of genes. Many of the encoded proteins are associated with the immune response, alongside a number of other cellular functions (reviewed in Verheijen & Sleegers, 2018).
The defining hallmark pathologies of AD are amyloid-β plaques and neurofibrillary tangles alongside substantial neurodegeneration. Amyloid-β plaques accumulate first and result from cleavage of APP into (among many other metabolites) amyloid-β-40 and amyloid-β-42, which abnormally fold and aggregate in the brain. Neurofibrillary tangles arise from hyperphosphorylation of tau protein, leading to neuronal damage and neurodegeneration (Selkoe & Hardy, 2016). Neuroinflammation is also a classic feature of the disease, involving reactive microglia and astrocytes. Indeed, microglia have been found to co-localize with plaques in postmortem human brain (Calsolaro & Edison, 2016) and in mouse models of AD (e.g., Medawar et al., 2019).
Clinical features of AD include initial problems with episodic memory and difficulties multitasking, followed by more profound cognitive impairment as the disease progresses, including behavioral and mood changes, and eventual decline into full dementia (Jost & Grossberg, 1995).
7.1 Structural abnormalities of the basal ganglia in Alzheimer's disease
The prevalence of AD undoubtedly makes it a crucial disease to find a cure for; however, much of the research to date has focused primarily on the hippocampus and the neocortex. The basal ganglia have received little attention in this context, despite evidence of degeneration of structures in the deep gray matter which may contribute to AD symptoms (de Jong et al., 2008). Early evidence from Braak and Braak (1991b) indicated that, in cases of severe AD, there are large numbers of extracellular amyloid-β deposits in all thalamic nuclei and severe changes in the limbic nuclei due to the presence of neurofibrillary tangles, indicating an impairment of the limbic circuits (Braak & Braak, 1991a).
Continuing research has consistently demonstrated that AD patients have shape changes in the basal ganglia, aided by recent advances in brain imaging techniques. Patients exhibit more rapid shape changes in the thalamus and basal ganglia compared with age-matched controls (Cho et al., 2014); the medial heads on the caudate nucleus and ventral lateral putamen are selectively degenerated (de Jong et al., 2011); and MRI indicates that volumes of caudate nucleus (Almeida et al., 2003), putamen, and thalamus (de Jong et al., 2011) are reduced in AD patients. Moreover, in all cases, the structural changes were correlated with cognitive decline. Thus, it is clear that, contrary to the standard declaration that the basal ganglia and associated subcortical structures are spared in AD, these regions are susceptible to structural changes during disease progression and may well underlie aspects of cognitive impairment.
The striatum, comprising of the nucleus accumbens, caudate nucleus, and putamen, appears to be particularly vulnerable in AD. Indeed, both diffuse plaques and neurofibrillary tangles are observed in striatal areas and amyloid-β deposits are found in the striatum in young, asymptomatic members of families with PSEN1 mutations (Klunk et al., 2007). Additionally, amyloid-β levels may increase in the basal ganglia over 10 years before expected symptom onset (Bateman et al., 2012), suggesting that it may be a brain region affected early in the disease and perhaps an underexplored therapeutic target.
As the basal ganglia are believed to drive higher level cognitive processes such as attention and memory, it is perhaps unsurprising that degeneration and shape changes would affect cognitive function in AD. Little, however, is known about this, as robust studies correlating alterations in subcortical volumes in AD with cognitive function are lacking.
7.2 Iron-associated basal ganglia dysfunction in Alzheimer's disease
Metabolic processing is known to dysregulate with age and is particularly affected in neurodegeneration. The basal ganglia (in particular the putamen, caudate nucleus, and globus pallidus) have a high metabolic rate and contain the highest levels of iron in the brain (Ramos et al., 2014), which may render them particularly sensitive to age-associated metabolic dysregulation. Even in the healthy aging brain, iron accumulates in cells leading to a region-specific increase in total iron levels (Connor, Menzies, St Martin, & Mufson, 1990). As aging represents the most significant risk factor for neurodegenerative diseases, iron may be involved as either a cause or a consequence. This is evidenced by studies showing that iron accumulation in the basal ganglia is a common feature of many neurodegenerative diseases and results in both cognitive and motor dysfunction. For example, iron has been found to co-localize with the insoluble protein aggregates that characterize neurodegenerative diseases such as AD, PD, HD, and DLB (Agrawal, Fox, Thyagarajan, & Fox, 2018; Joppe, Roser, Maass, & Lingor, 2019; van Bergen et al., 2016), suggesting a dysfunction in iron trafficking or metabolism as a cause or consequence of pathology. The heavy iron load in the basal ganglia may explain its increased susceptibility to neurodegeneration, so this represents an important brain region to further examine in this context.
Certain disorders are characterized by iron dysregulation (Dusek, Jankovic, & Le, 2012) but the impact of this in AD is not fully understood. An early study of the basal ganglia in AD revealed that patients who have basal ganglia mineralization have a higher rate of psychotic and motor disturbances, such as delusions, depression, parkinsonism and myoclonus, compared with age- and gender-matched AD patients without mineralization in the basal ganglia (Forstl, Burns, Cairns, Luthert, & Levy, 1992). Autopsy studies have shown that the highest concentrations of iron in the brain exist in the basal ganglia including the globus pallidus, putamen, and substantia nigra (Conway et al., 1998). Amyloid-β deposition and aggregation has been shown to be accelerated by metal ions such as iron and zinc (Lovell, Robertson, Teesdale, Campbell, & Markesbery, 1998) and iron has been demonstrated in vitro to increase the aggregation and cytotoxicity of amyloid-β peptide (Galante, Cavallo, Perico, & D'Arrigo, 2018). Iron also appears to be involved with plaques and tangles, the two hallmark pathologies of AD. Amyloid-β plaques co-localize with iron (van Bergen et al., 2016), suggesting that iron may either exacerbate amyloid-β aggregation or that the plaques may trap iron in some way. In mouse models, iron has been demonstrated to correlate with amyloid-β plaque morphology, with evidence for the formation of an iron–amyloid complex (Telling et al., 2017). Iron has also been found to bind to tau protein, inducing phosphorylation and aggregation (Liu, Fan, Yang, Wang, & Guo, 2018), thereby promoting the accumulation of hyperphosphorylated tau which leads to neurofibrillary tangles. Neuropathological studies appear to support this idea as iron levels in eight regions of the AD brain, including the globus pallidus and caudate nucleus, are raised compared to healthy controls, implicating the basal ganglia as part of the pathological profile of AD (Tao, Wang, Rogers, & Wang, 2014). It appears iron metabolism is impaired in some way in AD and other neurodegenerative diseases but how this relates to the basal ganglia or precisely to disease progression requires further studies. An improved understanding of this would aid the development of treatment strategies, such as developing iron chelators to sequester excess iron in the brain.
8 DEMENTIA WITH LEWY BODIES
DLB is the second most common form of dementia and occurs in approximately 15 out of 100 patients with dementia, when diagnosed by autopsy (McKeith et al., 2005) and accounts for 4–8% clinically (Vann Jones & O'Brien, 2014). Clinically, the disease is characterized by increasing deficits in executive, visual–spatial, and memory functions (Lippa et al., 2007). Diagnostically, the core features of the disease are fluctuation in cognition and attention (especially between periods of lucidity and confusion), recurrent visual hallucinations, and rapid eye movement sleep behavior disorder that precede or occur within a year of parkinsonian motor symptoms (Walker, Possin, Boeve, & Aarsland, 2015). This is highly similar to a condition called Parkinson's disease dementia; however, dementia in Parkinson's disease dementia is diagnosed only when it occurs in the presence of long-established motor symptoms (Jellinger & Korczyn, 2018). Indeed, the pathological features of both disorders are also highly similar (Jellinger, 2018). This disorder is frequently misdiagnosed due to its similarity of symptoms with AD; however, the presence of visual hallucinations is a distinguishing factor (Thomas et al., 2018).
Like PD, both DLB and Parkinson's disease dementia are classified as Lewy body disorders. Lewy body disorders are characterized by the presence of protein aggregates predominantly in the soma (Lewy bodies) or processes (Lewy neurites) of neurons (Braak, Ghebremedhin, Rub, Bratzke, & Tredici, 2004; Forno, 1996). Lewy bodies measure 8–30 μm in diameter with a spherical shape, with or without a dense granular core and a periphery of radiating filaments, which appear pink with a typical hematoxylin and eosin staining (Olanow, Perl, DeMartino, & McNaught, 2004). In the brainstem, Lewy bodies possess a densely stained core and a lightly stained peripheral halo, while in the cortex Lewy bodies lack the halo (Forno, 1996; Kosaka, Oyanagi, Matsushita, Hori, & Iwase, 1976). The inner core stains more intensely for α-synuclein, while the periphery stains more strongly for ubiquitin, with the α-synuclein staining being more specific (Spillantini et al., 1997). Approximately two thirds of Lewy bodies in the substantia nigra stain for α-synuclein and approximately one third in the cortex (Sakamoto et al., 2002). More than 76 other proteins have been found in Lewy bodies and neurites (Wakabayashi, Tanji, Mori, & Takahashi, 2007).
Mutations in the α-synuclein gene (SNCA) are linked to familial cases in PD and DLB (Polymeropoulos et al., 1997; Zarranz et al., 2004). Genes linked to both AD and PD are also linked to DLB, for example, GBA and APOE genes (Guerreiro et al., 2018; Nalls et al., 2013; Tsuang et al., 2012). Mutations in other genes linked to PD, such as LRRK2 and SCARB2, may also play a role in disease risk (Alcalay et al., 2016; Bras et al., 2014; Hyun, Yoon, Lee, & Lee, 2013; Orme, Guerreiro, & Bras, 2018). A recent review of the genetics of DLB can be found in Guerreiro et al. (2019).
α-Synuclein is highly expressed in the brain, located in pre- and postsynaptic terminals, and may make up as much as 1% of the cytosol (Kim, Kagedal, & Halliday, 2014). It is a 140 amino acid-long typically unfolded protein, with an amino-terminal lipid-binding α-helix, a non-amyloidogenic core domain, and an unstructured carboxy-terminal. Although its exact physiological role is unclear, α-synuclein likely has a role in neurotransmitter release, potentially by regulating the exocytosis of vesicles by interactions with the SNARE complex (Murphy, Rueter, Trojanowski, & Lee, 2000; Orme et al., 2018). When α-synuclein becomes misfolded, the random coil of the non-amyloidogenic core domain forms β-sheets, which are important for protofibril and fibril formation (Serpell, Berriman, Jakes, Goedert, & Crowther, 2000; Uversky & Eliezer, 2009). The spread of Lewy bodies in a rostrocaudal direction in PD suggests a prion-like transmission (Braak et al., 2004; George, Rey, Reichenbach, Steiner, & Brundin, 2013; Seidel et al., 2015). Strong evidence for this spread comes from studies in which embryonic neurons grafted into PD patients to replace lost nigral cells revealed α-synuclein/Lewy body pathology upon postmortem examination (Kordower, Chu, Hauser, Freeman, & Olanow, 2008; Kurowska et al., 2011; Li et al., 2008). Furthermore, propagation of Lewy bodies has been shown in animal and cell culture models (Karpowicz, Trojanowski, & Lee, 2019; Kordower et al., 2011; Ngolab et al., 2017).
8.1 Pathology
Lewy bodies have been linked to cell death and the extent of Lewy body pathology correlates with clinical pathology in both DLB (Tiraboschi et al., 2015) and PD with dementia (Hurtig et al., 2000). Lewy body pathology is found widespread in postmortem brains from both groups of patients, especially affecting the frontal cortex, midbrain and brainstem nuclei, dorsal efferent nucleus of the vagus nerve, basal forebrain nuclei, and limbic regions (Jellinger & Korczyn, 2018; Neef & Walling, 2006). This widespread pathology makes the spreading of the disorder more difficult to define than in PD, as it does not appear to extend in a rostro-caudal manner (Walker et al., 2015), although systems to stage the disorder have been proposed (McKeith et al., 2005).
Gray matter atrophy occurs in frontotemporal, occipital and parietal cortical areas and subcortical atrophy of amygdala, caudate, putamen, and pallidum (Almeida et al., 2003; Beyer, Domingo-Sabat, & Ariza, 2009; Gazzina et al., 2016; Sanchez-Castaneda et al., 2009; Watson, O'Brien, Barber, & Blamire, 2012). Like PD, dopaminergic and cholinergic neurons appear especially vulnerable in the disease and in some cases have been found to be more degenerated than in PD (Colloby, McParland, O'Brien, & Attems, 2012; Muenter et al., 1998; Outeiro et al., 2019; Thomas et al., 2017). Dopamine synthesis, transporter binding, and storage are all decreased in patients with DLB (Gomperts et al., 2016; Marquie et al., 2014; Nikolaus, Antke, & Muller, 2009). Cholinergic deficits in the cortex are observed due to the loss of neurons in the basal forebrain, leading to cholinergic denervation and signaling deficits (Grothe et al., 2014; Mazere, Lamare, Allard, Fernandez, & Mayo, 2017).
Cell death due to an impairment of axonal transport, autophagy–lysosomal, ubiquitin–proteasome, and mitochondrial pathways have all been strongly implicated in the disease (Manecka, Vanderperre, Fon, & Durcan, 2017; Power, Barnes, & Chegini, 2017; Sato et al., 2018; Volpicelli-Daley, 2017). Importantly, whether this is directly due to Lewy bodies is unclear, as they may play a role in protecting the cell through the uptake of misfolded and nonfunctional proteins (Beyer et al., 2009), as there are cases of individuals with a significant Lewy body pathology at autopsy that do not demonstrate symptoms of dementia (Parkkinen, Pirttila, & Alafuzoff, 2008). Oligomers of α-synuclein may also mediate the cellular toxicity in the disease (Outeiro et al., 2019). An additional factor that may play a role in cell death is the co-occurrence of AD pathology in patients with DLB, which is suggested to be an increasingly important part of the disorder rather than a comorbidity (Deramecourt et al., 2006; Jellinger & Korczyn, 2018). This may lead to synergistic effects and increased cell death. Indeed, AD and DLB co-pathologies have been shown to lead to a greater temporal lobe atrophy (van der Zande et al., 2018) and greater baseline amyloid-β imaging predicts more severe neurodegeneration in DLB patients (Sarro et al., 2016).
9 CONCLUDING REMARKS
The evidence that the basal ganglia are implicated in dementia is limited but, particularly in a subset of neurodegenerative diseases, it is becoming ever more apparent that their role is not restricted to motor control. As the physiological roles of the basal ganglia and associated structures in cognition and other higher functions become better appreciated, the dysfunction associated with their pathology is broadening. Whether targeting the basal ganglia for treatments of late stages of the less prominent disorders of the basal ganglia, such as AD, is a worthwhile approach is yet to be ascertained but understanding how they are affected in the disease may indeed reveal disease-modifying targets.
CONFLICT OF INTERESTS
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
Conceptualization, D.M.C.; Writing—Original Draft, K.S.V., K.M.S., D.P.B., and J.B.; Writing—Review & Editing, K.S.V. and D.M.C.; Supervision, D.M.C.