

Resistin protects against 6-hydroxydopamine-induced cell death in dopaminergic-like MES23.5 cells
Abstract
Resistin is originally reported as an adipose tissue-specific hormone and is thought to represent a link between obesity and insulin-resistant diabetes. Adipokines exert energy-regulation and has been reported to have neuroprotective effect like leptin, adiponectin, and ghrelin. However, the role of resistin in neuroprotective effect has not been explored. 6-hydroxydopamine (6-OHDA), one of the most investigated Parkinson's disease neurotoxins, is widely used to study mechanisms of cell death in dopaminergic neurons. In the present study, our results show that treatment of resistin protects 6-OHDA-induced cell death in dopaminergic-like MES23.5 cells. Resistin also antagonizes 6-OHDA-induced apoptotic cell death measured by fluorescence-activated cell sorter (FACS) analysis and Hochest 33342 staining. Furthermore, treatment of resistin also dramatically reduces 6-OHDA-mediated ROS production and mitochondria transmembrane potential dissipation. Moreover, expression of 6-OHDA-induced apoptotic markers, such as Bcl-2 degradation, Bax expression, PARP degradation and caspase 3 activity increase, are all attenuated by resistin treatment. Our results also show that resistin induces up-regulation of heat shock protein (Hsp) 32 (heme oxygenase-1, HO-1) and Hsc (heat shock cognate) 70. The protective effect of resistin on 6-OHDA-induced cell death is abolished by HO-1 inhibitor zinc protoporphyrin IX and HSP inhibitor KNK437. These results suggest the neuroprotective effects of resistin against 6-OHDA-induced cell death with the underlying mechanisms of inhibiting oxidative stress and apoptosis. Therefore, we suggest that resistin may provide a useful therapeutic strategy for neurodegenerative diseases such as Parkinson's disease. J. Cell. Physiol. 228: 563–571, 2013. © 2012 Wiley Periodicals, Inc.
Adipocytes can produce many adipokines, including leptin, adiponectin, resistin, TNF-α, and IL-6 (Tschop et al., 2001). It has been shown that circulating peptides can gain access to the central nervous system either by simple lipophilic diffusion or by saturable transport systems, such as the case of leptin and ghrelin (Banks et al., 2002; Pan et al., 2006). Adipokines are well known for the function of regulating energy balance (Arner, 2005), some of them have also been proved to be neuroprotective, such as leptin (Wang et al., 2010; Perez-Gonzalez et al., 2011) and adiponectin (Qiu et al., 2011). Moreover, ghrelin, a peptide secreted mainly by stomach, can increase appetite. However, ghrelin also has been reported to exert neuroprotective effects (Moon et al., 2009; Lee et al., 2010). Resistin belongs to a family of cysteine-rich proteins and each with a unique tissue distribution (Steppan et al., 2001a, b). In rodents, resistin is predominantly expressed in white adipose tissue (Holcomb et al., 2000; Steppan et al., 2001b), and resistin was suggested to be a link between obesity and diabetes (Steppan et al., 2001a). Since resistin is secreted by adipose tissue and presents in the circulation, it is presumed to act at distant tissues (Banerjee and Lazar, 2003). Importantly, it has been reported that leptin and ghrelin protect dopaminergic neuronal cell death (Lu et al., 2006; Moon et al., 2009), however, the effects of resistin in neuronal cells remains unclear.
Parkinson's disease (PD) is characterized by the progressive degeneration of dopaminergic neurons in the substantia nigra, giving dopamine depletion in the striatum. The resulting loss of dopamine in the striatum leads to debilitating motor dysfunction including rigidity, resting tremor, mask face, and bradykinesia. Although the exact causes of PD are not clear yet, oxidative stress, mitochondrial dysfunction, and environmental factors, genetic mutations are considered to be involved. Accumulating evidence supports that among the proposed mechanisms of dopaminergic degeneration, oxidative stress plays an important role in cell death in PD (Burkhardt and Weber, 1994; Bostanci and Bagirici, 2008; Drechsel and Patel, 2009). Apoptosis has been implicated as the important mechanism leading to dopaminergic neuronal cell death in PD (Bredesen et al., 2006; Jenner and Olanow, 2006). Therefore, anti-apoptosis strategies could prevent or delay the progress of PD. Previous report has shown that ghrelin protects MPTP-induced neurotoxicity to the dopaminergic neurons in SN (Jiang et al., 2008). 6-hydroxydopamine (6-OHDA), a neurotoxin in PD experimental model, inhibits complex I of the mitochondrial respiratory chain, and induces oxidative stress and dopaminergic neurodegeneration (Youdim et al., 2001; Guo et al., 2005). In the present study, we conducted our experiment in 6-OHDA-induced neuronal cell death in MES23.5. The results showed that resistin can abolish 6-OHDA-induced apoptosis in MES23.5 cells. The protective effect of resistin involves the up-regulation of HO-1 and Hsc70 expression.
Materials and Methods
Reagents
Resistin and adiponectin were purchased from PeproTech (Rocky Hill, NJ). Fetal bovine serum (FBS), Lipofectamine™ 2000 (LF2000), Dulbecco's modified Eagle's medium (DMEM), DMEM/F12 and OPTI-MEM were purchased from Gibco BRL (Invitrogen Life Technologies, Carlsbad, CA). Goat anti-mouse and anti-rabbit horseradish peroxidase-conjugated IgG, primary antibodies against Bax, Bcl-2, β-actin, and PARP1/2 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Primary antibody against cleavage caspase-3 was purchased from Cell Signaling and Neuroscience (Danvers, MA). Primary antibodies against Hsc70 and HO-1 were purchased from StressGen Biotechnologies (Victoria, BC, Canada). Primary antibody against HSF1 was purchased from Enzo Life Sciences (Farmingdale, NY). ZnPPIX and KNK 437 were purchased from Calbiochem (San Diego, CA). Leptin and other chemicals were obtained from Sigma–Aldrich (St. Louis, MO).
Cell cultures
MES23.5 cells (Dr. W.-D. Le, Baylor College of Medicine, TX) is a dopaminergic cell line hybridized from murine neuroblastoma-glioma N18TG2 cells with rat mesencephalic neurons, which exhibits several properties similar to the primary neurons originated in the substantia nigra (Crawford et al., 1992). The cells were cultured in DMEM/F12 containing Sato's components growth medium supplemented with 5% FBS, 100 units/ml of penicillin and 100 mg/ml of streptomycin at 37°C, in a humid 5% CO2, 95% air environment (Lu et al., 2010).
Hoechst 33258 staining
Hoechst 33258 is a DNA-binding fluorescent dye and used to determine apoptotic nuclei. Cells were incubated with resistin for 1 h, followed by the treatment with 6-OHDA for 8 h and then stained with Hoechst 33258 (1 µg/ml) for 10 min. Results were determined by visual observation of nuclear morphology through fluorescence microscopy. Nuclear shrinkage, chromatin condensation, and nuclear fragmentation were considered as apoptosis.
MTT assay
Cell viability was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. After treatment with 6-OHDA for 24 h, cultures were washed with PBS. MTT (0.5 mg/ml) was then added to each well and the mixture was incubated for 1 h at 37°C. Culture medium was washed with PBS and then replaced with DMSO to dissolve formazan crystals. After the mixture was shaken at room temperature for 10 min, absorbance was determined at 550 nm using a microplate reader (Bio-Tek, Winooski, VT).
Sulforhodamine B assay (SRB)
The SRB assay is prepared according to our previous report (Huang et al., 2011) and based on the measurement of cellular protein content. After treatment with 6-OHDA for 24 h, cells were fixed with 10% trichloroacetic acid and stained by 0.4% (w/v) SRB in 1% acetic acid for 30 min. Unbound SRB was washed out by 1% acetic acid and SRB-bound cells were resolved by Trizma base (10 mM). The absorbance was read at a wavelength of 515 nm using a microplate reader (Bio-Tek).
LDH assay
Secreted LDH in the medium was assayed with LDH kits (Promega, Madison, WI) according to the manufacturer's procedures. The supernatant was incubated with the LDH assay reaction mixture in the dark at 37°C for 30 min. The absorbance was read at a wavelength of 450 nm using a microplate reader (Bio-Tek).
Quantification of apoptosis by flow cytometry
Apoptosis was assessed by binding of Annexin V protein to exposed phosphatidylserine (PS) residues at the surface of cells undergoing apoptosis as previously described (Liu et al., in press; Lu et al., 2012). Cells were treated with vehicle or 6-OHDA for 24 h and then washed twice with PBS and re-suspended in staining buffer containing propidium iodide (PI, 1 µg/ml) and Annexin V-FITC (0.025 µg/ml). Double-labeling was performed at room temperature for 10 min in darkness before flow cytometric analysis. Cells were immediately analyzed using FACScan and the Cellquest program (Becton Dickinson, Lincoln Park, NJ).
Western blotting
Cells were plated in six-well plates for 24 h and treated with resistin for various concentrations and then washed with cold PBS, laid on ice with radioimmunoprecipitation assay buffer for 30 min. Protein concentrations were determined by colorimetric assay using Bio-Rad assay kit (Bio-Rad, Hercules, CA). Samples containing 40 µg protein were separated on sodium dodecyl sulphate (SDS)–polyacrylamide gels and transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA). The membranes were incubated with 5% dry skim milk in PBS for 1 h to block nonspecific binding. Following incubation with primary antibodies overnight at 4°C, the membranes were then incubated with secondary antibody for 1 h. The blots were visualized by enhanced chemiluminescence (Millipore, Bedford, MA) using Kodak X-OMAT LS film (Eastman Kodak, Rochester, NY).
RT-PCR
Total RNA was extracted from MES23.5 cell line using a TRIzol kit (MDBio, Inc., Taipei, Taiwan). The reverse transcription reaction was performed to reverse transcribe 2 µg of total RNA into cDNA using oligo(dT) primer. After preincubation at 50°C for 2 min and 95°C for 10 min, the PCR was performed for 30 cycles of 95°C for 10 s and 60°C for 1 min. The threshold was set above the non-template control background and within the linear phase of target gene amplification to calculate the cycle number at which the transcript was detected (denoted as CT). The oligonucleotide primers were HO-1: 5′-TCTATCGTGCTCGCATGAAC-3′ and 5′-CAGCTCCTCAAACAGCTCAA-3′; Hsc70: 5′-CCAGGTGTGATCTAGGAGAC -3′ and 5′- GTAACACACCTCTTGCTAT-3′; and GAPDH: 5′-ACCACAGTCCATGCCATCAC-3′ and 5′-TCCACCACCCTGTTGCTGTA-3′.
Reactive oxygen species (ROS) assay
The production of intracellular O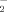 and hydrogen peroxide (H2O2) were assessed spectrofluorimetrically by oxidation of specific probes dihydroethidium (DHE) and 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA), respectivily (Sigma–Aldrich). Cells were plated at 6 well-plate and exposed to 6-OHDA for another 2 h. The cells were incubated with DHE (10 µM) or H2DCFDA (10 µM) for 30 min at 37°C. The fluorescence intensity was measured with excitation filter of 488 and 525 nm emission wavelengths using the flow cytometry.
and hydrogen peroxide (H2O2) were assessed spectrofluorimetrically by oxidation of specific probes dihydroethidium (DHE) and 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA), respectivily (Sigma–Aldrich). Cells were plated at 6 well-plate and exposed to 6-OHDA for another 2 h. The cells were incubated with DHE (10 µM) or H2DCFDA (10 µM) for 30 min at 37°C. The fluorescence intensity was measured with excitation filter of 488 and 525 nm emission wavelengths using the flow cytometry.
Mitochondrial transmembrane potential
Mitochondrial activity was assessed using the fluorescent probe, rhodamine123 dye (Chiou et al., 2012). The accumulation of rhodamine123 in mitochondria depends on mitochondrial transmembrane potential. Cells were plated at a 24 well-plate and incubated with 6-OHDA for 2 h. Cells were incubated with rhodamine123 (10 µM) for 30 min at 37°C and then washed twice with PBS. Staining results were visualized with fluorescence microscope (Nikon, Tokyo, Japan). The fluorescence intensity was excited by 488 nm laser light and emission was captured at 530 nm.
Electrophoretic mobility shift assay (EMSA)
The EMSA gel shift assay was performed according to the manufacturer's protocol (Panomics, Redwood City, CA). Briefly, nuclear extract of cells was incubated with poly d(I-C), and then incubated with biotin-labeled probes for 30 min. The double stranded sequence of oligonucleotides containing the HSE was 5′-CTAGAAGCTTCTAGAAGCTTCTAG-3′ (Ethridge et al., 1998). After electrophoresis on an 8% polyacrylamide gel, the samples on gel were transferred onto a presoaked Immobilon-Nyt membrane (Millipore, Billerica, MA). The membrane was cross-linked in an oven and then developed by the blocking buffer and streptavidin–horseradish peroxidase conjugate, before being subjected to Western blot analysis.
Statistical analysis
Statistical analysis was performed using software Graphpad Prism 4.01 (Graph Pad Software, Inc., San Diego, CA). Values are means ± SEM. Statistical analysis of the difference between two samples was performed using Student's t-test. In all cases, a value of P < 0.05 was considered to be significant.
Results
Resistin attenuates 6-OHDA-induced cell death and morphological damage
As shown in Figure 1, after the exposure to 75 µM 6-OHDA for 24 h, the cell viability declined significantly to 43.10 ± 3.91%. However, its cytotoxic effects were ameliorated in the pretreatment of resistin. The results from MTT (Fig. 1A), SRB (Fig. 1B), and LDH (Fig. 1C) assays indicated that resistin exhibited protective effect against the damage caused by 6-OHDA. The morphological change visualized by phase-contrast imaging showed that the cell bodies were evenly attached with regular shape in the control group. Shrinkage and detachment were observed in the cells treated with 6-OHDA for 24 h. Pretreatment with resistin significantly reversed the morphological damage caused by 6-OHDA (Fig. 1D). In addition, treatment of resistin alone did not have significant effect on the cell viability and morphology compared with the control.

Resistin attenuated 6-OHDA-induced cell death and morphological damage. Cells were preincubated with resistin at various concentrations for 1 h and then 6-OHDA (75 µM) was added for an additional 24 h and cell viability was analyzed by MTT assay (A), SRB assay (B), and LDH assay (C). Results are expressed as means ± SEM of four independent experiments. *, P < 0.05, as compared to the control group; #, P < 0.05, as compared to the 6-OHDA-treated group. D: Cells were pretreated with resistin (5 ng/ml) for 1 h and 6-OHDA (75 µM) was added for an additional 24 h. The morphological change was visualized by phase-contract imaging. Scale bar: 50 µm.
Resistin reverses 6-OHDA-induced apoptosis
Nuclear condensation and DNA fragmentation are hallmarks of cell apoptosis. In this study, we investigated the protective effect of resistin on 6-OHDA-induced nuclear condensation and DNA fragmentation by Hoechst 33258 staining. As shown in Figure 2A, 6-OHDA treatment alone for 8 h induced obvious nuclear condensation, fragmentation, and size reduction compared with the control. All these characteristics of apoptosis were decreased significantly when cells were pre-incubated with resistin. It has been reported that 6-OHDA treatment resulted in annexin V positive in dopaminergic neuronal cell death (Lotharius et al., 1999; Ikeda et al., 2008). As compared to vehicle-treated cells, 6-OHDA treatment alone induced cell apoptosis. Pretreatment with resistin reversed apoptosis caused by 6-OHDA (Fig. 2B) at 24 h. Importantly, our results also observed resistin reduces annexin V-positive reaction at early apoptosis (Fig. 2C). 6-OHDA-induced apoptotic cells at early apoptosis reveals annexin V-positive but PI-negative reaction (Yoon et al., 2009). As shown in Figure 2C, resistin reduced 6-OHDA-medicated annexin V-positive reaction of early phase apoptosis at 12 h as well.

Effect of resistin on 6-OHDA-induced apoptosis in MES23.5 cells. A: Cells were pretreated with resistin (5 ng/ml) for 1 h and 6-OHDA (75 µM) was added for an additional 8 h. The Hochest staining was visualized by florescence imaging. 6-OHDA treatment alone for 8 h induced obvious nuclear condensation and fragmentation indicated by arrows. Scale bar: 10 µm. B: Cells were incubated in vehicle, 6-OHDA (75 µM) or resistin (5 ng/ml) for 24 h; or cells were preincubated with various concentrations (2, 5, or 10 ng/ml) of resistin and then exposed to 6-OHDA for 24 h. C: cells were preincubated with resistin (5 ng/ml) and then exposed to 6-OHDA for 12 h. The percentage of apoptotic cells was analyzed by flow cytometry of Annexin V/PI double staining.
Resistin inhibites 6-OHDA-induced elevation in intracellular ROS levels
Since ROS elevation initiates all the pathogenesis process induced by 6-OHDA, we then investigated the intracellular O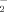 and H2O2 formation using a fluorescent sensitive probe DHE and DCFH-DA, and were determined by flow cytometry. As shown in Figure 3, 6-OHDA induced a significant increase of DHE and DCFH-DA fluorescence, reflecting the increase of ROS. 6-OHDA treatment alone for 2 h induced ∼2.8- and 5.7-fold increase in O
and H2O2 formation using a fluorescent sensitive probe DHE and DCFH-DA, and were determined by flow cytometry. As shown in Figure 3, 6-OHDA induced a significant increase of DHE and DCFH-DA fluorescence, reflecting the increase of ROS. 6-OHDA treatment alone for 2 h induced ∼2.8- and 5.7-fold increase in O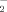 and H2O2 levels, respectively. However, pretreatment with resistin for 1 h concentration-dependently decreased the O
and H2O2 levels, respectively. However, pretreatment with resistin for 1 h concentration-dependently decreased the O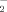 (Fig. 3A) and H2O2 (Fig. 3B) levels.
(Fig. 3A) and H2O2 (Fig. 3B) levels.

Resistin reduced 6-OHDA-induced ROS generation in MES23.5 cells. O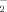 and H2O2 generation were determined using the fluorescence probes DHE (A) and DCFH-DA (B), respectively. Cells were pretreated with resistin (5 ng/ml) for 1 h and 6-OHDA (75 µM) was added for an additional 2 h, the production of O
and H2O2 generation were determined using the fluorescence probes DHE (A) and DCFH-DA (B), respectively. Cells were pretreated with resistin (5 ng/ml) for 1 h and 6-OHDA (75 µM) was added for an additional 2 h, the production of O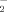 and H2O2 were examined by flow cytometry. Results are expressed as the mean ± SEM of four independent experiments. *, P < 0.05, as compared to the control group; #, P < 0.05, as compared to the resistin-treated group.
and H2O2 were examined by flow cytometry. Results are expressed as the mean ± SEM of four independent experiments. *, P < 0.05, as compared to the control group; #, P < 0.05, as compared to the resistin-treated group.
Resistin reverses mitochondrial abnormalities induced by 6-OHDA
It is generally accepted that the 6-OHDA-induced neuronal apoptosis is mediated by the mitochondrial dysfunction characterized by several events (Bueler, 2010). Therefore, we investigated whether resistin could restore the impaired mitochondrial function. ΔΨm was measured using fluorescent probes rhodamine123. The results shown in Figure 4 revealed that 6-OHDA treatment alone decreased rhodamine123 fluorescence, reflecting the decrease in ΔΨm. Resistin pretreatment significantly reversed the decrease in ΔΨm in a concentration-dependent manner. Additionally, resistin treatment alone had no apparent effect on ΔΨm compared with the control.

Resistin restored 6-OHDA-induced decrease in ΔΨm in MES23.5 cells. Mitochondria membrane potential was determined using a fluorescence probe rhodamin 123. A: Cells were pretreated with resistin (5 ng/ml) for 1 h and 6-OHDA (75 µM) was added for an additional 2 h. Representative pictures were taken by a fluorescence microscope (20×). The experiments were repeated three times independently, with one representative result shown. Note that the levels of ΔΨm in 6-OHDA-treated cells were significantly decreased compared with control, while cells pretreated with resistin showed a relatively high level of ΔΨm. B: Cells were pretreated with various concentrations of resistin for 1 h and 6-OHDA (75 µM) was added for an additional 2 h, the ΔΨm levels were examined by flow cytometry. Results are expressed as the mean ± SEM of four independent experiments. *, P < 0.05, as compared to the control group; #, P < 0.05, as compared to the resistin-treated group.
Resistin restores the Bax/Bcl-2 ratio and prevented PARP and caspase-3 activation
The mitochondrial damage results in release of apoptotic proteins and triggers apoptosis of cells. Bax and Bcl-2, the two main members of Bcl-2 family, can affect mitochondrial membrane permeability (Vander Heiden and Thompson, 1999). Caspase-3 is a key protein regulator of apoptosis (Budihardjo et al., 1999). Next, we examine the expression of Bax, Bcl-2, and cleaved caspase-3 after resistin treatment. 6-OHDA treatment caused a dramatic decrease of the anti-apoptotic protein Bcl-2 expression (Fig. 5A), and a significant increase of the pro-apoptotic protein Bax (Fig. 5B). Moreover, pretreatment with resistin concentration-dependently reversed the Bax/Bcl-2 ratio in MES23.5 cells (Fig. 5A,B). The expression of activated PARP-1/2 and cleaved caspase-3 were both increased after exposure to 6-OHDA. Pretreatment with resistin concentration-dependently reversed these changes (Fig. 5C,D). In addition, treatment with resistin did not affect apoptotic protein expression (Supplementary Fig. S2A).

Resistin increases Bax, Bcl-2, PARP, and cleaved caspase-3 expression. Cells were pretreated with various concentrations of resistin for 1 h, followed by exposure to 6-OHDA (75 µM) for 24 h, the protein levels of Bax, Bcl-2, PARP, and cleaved caspase-3 were examined by Western blot analysis. Results are the representative of three independent experiments.
Protective effect of heat shock proteins in 6-OHDA-induced cell death
It has been reported that Hsc70 (Li et al., 1996; Creagh and Cotter, 1999; Koul et al., 2008) and HO-1 (Rieder et al., 2004) exert neuroprotective effect. Here, we examine whether resistin regulates Hsc70 and HO-1 expression. Resistin increased Hsc73 expression in a concentration- (Fig. 6A) and a time- (Fig. 6D) dependent manners. On the other hand, resistin also dramatically increased HO-1 expression (Fig. 6B,E). Interestingly, we also found resistin increased Bcl-2 expression (Fig. 6C,F). Furthermore, we used HO-1 inhibitor ZnPPIX and HSP inhibitor KNK437 to evaluate the cell protective effect after 6-OHDA treatment. Pretreatment of ZnPPIX effectively reversed the protective effect of resistin determined by MTT (Fig. 7A), SRB (Fig. 7B), and LDH assay (Fig. 7C). On the other hand, pretreatment of KNK 437 effectively reversed the protective effect of resistin (Fig. 7D–F). Otherwise, treatment of ZnPPIX or KNK 437 alone did not have significant effect.

Resistin induces Hsc70, HO-1, and Bcl-2 up-regulation in MES23.5 cells. Cells were incubated with various concentrations of resistin for 24 h, the HSP73 (A), HO-1 (B), and Bcl-2 (C) expression were examined by Western blot analysis. Cells were incubated with resistin (5 ng/ml) for indicated time periods, the Hsc70 (D), HO-1 (E), and Bcl-2 (F) expression were examined by Western blot analysis. Results are the representative of three independent experiments.

Resistin protects 6-OHDA-induced cell death through heat shock protein expression. Cells were incubated with various concentrations of ZnPP IX (0.1 or 0.3 µM) or KNK 437 (25 or 50 µM) for 30 min followed by treatment with resistin (5 ng/ml) for 1 h and 6-OHDA for an additional 24 h. Cell viability was analyzed by MTT assay (A,D), SRB assay (B,E), and LDH assay (C,F). Treatment of ZnPP IX (0.3 µM) and KNK 437 (50 µM) at these dosages did not have significant effect. *, P < 0.05, as compared to the control group; #, P < 0.05, as compared to the 6-OHDA-treated group.
These results indicate that resistin effectively prevents 6-OHDA-induced ROS production, mitochondria abnormalities, PARP cleavage and caspase 3 activation. Resistin also up-regulates Hsc70 and HO-1 expression which results in neuroprotection.
Discussion
Increasing evidence has shown that numerous endogenous peptides such as leptin (Weng et al., 2007), ghelin (Andrews et al., 2009; Moon et al., 2009), and adiponectin (Qiu et al., 2011) exert protective effects against neurotoxin-induced cell death. However, the neuroprotective effects of resistin have not yet been reported. In the present study, we demonstrated that resistin could attenuate 6-OHDA-induced neurotoxicity through anti-oxidative mechanism in MES23.5 cells. This effect of resistin was achieved by the regulation of ROS levels, mitochondria function, and Bax/Bcl-2 ratio, as well as caspase-3 activity. The etiologic evidence proves that oxidative stress plays an important role in the onset and progression of PD, which could be blocked or delayed by a variety of antioxidants (de Vries et al., 2008; Kamat et al., 2008). Therefore, the possible neuroprotective effect of resistin on 6-OHDA-induced oxidative stress was first investigated in this study. Our results showed that 6-OHDA-treatment significantly increased ROS production, which could be reduced by resistin. This is consistent with other studies that the protective effect of adipokines ghrelin and leptin were possibly accomplished through the antioxidative activity (Lu et al., 2006; Weng et al., 2007; Jiang et al., 2008). Here, the combination of adipokines ghrelin, leptin, and resistin did not show synergic effect (Supplementary Fig. S4). Because our system is an in vitro model, the interactions of adipokines will need further investigations in animal model.
It has been reported that resistin is associated with inflammation in metabolic syndrome (Fargnoli et al., 2010; Kontunen et al., 2011), and might exert its effects via TLR-4 (Tarkowski et al., 2010; Jamaluddin et al., 2012). However, our results did not find the effect of resistin was regulated by TLR-4 receptor (Supplementary Fig. S1), suggesting the effect of resistin might depend on cell types and cellular events. The effective receptor and regulating inflammation of resistin in CNS will need further investigation. Recent report also showed that resistin signals through delta decorin in adipocyte progenitors (Daquinag et al., 2011). Although pharmacologic studies support correlation between resistin, obesity, and insulin resistance in mice, the role of resistin as a modulator in nervous system is not yet understood. It has also been reported that resistin protects against acute myocardial infarction (Gao et al., 2007). The present findings demonstrate that resistin protects 6-OHDA-induced dopaminergic neuronal cell death, and increases HO-1 and Hsc70 expression in MES23.5 cells. The protective effect of resistin on 6-OHDA-induced cell death is abolished by HO-1 and Hsp inhibitors. These results support the neuroprotective effects of resistin against 6-OHDA-induced cell death with the underlying mechanisms of inhibiting oxidative stress and apoptosis.
Heat shock proteins are highly conserved molecules involve in many essential cellular processes. Inducible Hsp70 family is a prominent heat shock protein and is a key component of the endogenous cellular stress response. In the CNS, Hsc70 expression increases under various injurious conditions such as hyperthermia, excitotoxicity, ischemia, and traumatic brain injury (Dutcher et al., 1998; Bertrand et al., 2000; Seidberg et al., 2003; Lai et al., 2005). Numerous reports have shown that Hsc70 plays an important role in anti-apoptosis and exerts a strong neuroprotective function (Li et al., 1996; Creagh and Cotter, 1999; Koul et al., 2008). HO-1 (Hsp 32) is also an inducible form found in large quantity in the brain or other tissues and can be rapidly induced by various oxidative-inducing agents (Platt and Nath, 1998; Le et al., 1999; Otterbein and Choi, 2000). Recently, we have reported that Omega-3 polyunsaturated fatty acids induce HO-1 expression and exert anti-neuroinflammatory responses in microglia (Lu et al., 2010). Previous reports have also shown that HO-1 plays a significant role in anti-apoptosis and anti-proliferation and exerts a strong neuroprotective function (Choi and Alam, 1996; Terry et al., 1998; Le et al., 1999; Lee and Chau, 2002; Minamino et al., 2001). Moreover, resistin also increases Hsp 60 and Hsp90 expression (Supplementary Fig. S2C). The neuroprotective effect of resistin exerts only by pretreatment, but not co-treatment or post-treatment with the neurotoxin (Supplementary Fig. S3). Although the effect of pretreatment has less value in clinical application, our finding is the first to demonstrate the molecular mechanism of resistin protecting 6-OHDA-induced neuronal cell death in dopaminergic neurons. Recently, we and other groups have reported that HO-1 (Lu et al., 2010; Chen et al., 2012) and Hsc70 (Marinova et al., 2009; Kong et al., 2011) expression through phosphatidylinositol 3-kinase/AKT signaling pathway. The transduction pathways activated by resistin will need further investigation.
Our results also revealed that resistin induced HO-1 and Hsc70 mRNA expression (Supplementary Fig. S2B), suggesting that resistin-increased heat shock protein expression may be regulated at transcriptional level. The current consensus in mammalian cells is that HSF (heat-shock transcription factor) is the most key regulator of the heat shock response with the remaining factors playing important roles in stress and development (Abravaya et al., 1992; McMillan et al., 1998; He et al., 2003). The primary level of heat-shock gene regulation occurs via transcriptional activation mediated by the translocation of the HSF to the nucleus, where they ultimately bind to the upstream promoter sequence heat-shock element (HSE; Abravaya et al., 1992). Inactive HSF-1 monomers exist in the cytoplasm; to induce an Hsp response, these monomers must translocate to the nucleus, bind to the promoter of the inducible Hsp, such as Hsp-70 (Wu et al., 1985). In presence study, we examined whether the HSF signaling is involved in resistin-regulated heat shock response. Treatment of resistin resulted in an accumulation of HSF1 in nucleus (Supplementary Fig. S5A). Moreover, stimulation of cells with resistin increased the DNA binding activity of HSE in nuclear extracts (Supplementary Fig. S5B). Our results suggest that resistin-regulated heat shock response may involve with HSF activation.
Taken together, our results demonstrated that resistin increases endogenous antioxidant protein Hsc70 and up-regulates HO-1 expression. Our study also indicated that when neuronal cells have adequate or enhanced levels of anti-oxidative agents, they can protect themselves from oxidative damage. Hence, explore the therapeutic potential of resistin may help us to identify target molecules for drug development and therapy against neurodegeneration.

