

TLR4-dependent induction of vascular adhesion molecule-1 in rheumatoid arthritis synovial fibroblasts: Roles of cytosolic phospholipase A2α/cyclooxygenase-2
Abstract
Lipopolysaccharide (LPS)/Toll-like receptor 4 (TLR4)-mediated signaling pathways have caught the attention of strategies designed for rheumatoid arthritis (RA). In this study, we identified that cPLA2α acted as a modulator of LPS-induced VCAM-1 expression and THP-1 (human acute monocytic leukemia cell line) adherence. Treatment of RA synovial fibroblasts (RASFs) with LPS, a TLR4 agonist, promoted the VCAM-1 expression and THP-1 adherence which were decreased by pretreatment with a selective cytosolic phospholipase A2 (cPLA2) inhibitor (AACOCF3), implying the involvement of cPLA2α in these responses. This notion was further confirmed by knockdown of cPLA2α expression by transfection with cPLA2α small interfering RNA (siRNA) leading to a decrease in VCAM-1 expression and THP-1 adherence induced by LPS. Subsequently, the LPS-stimulated cPLA2α phosphorylation was attenuated by pretreatment with a MEK1/2 inhibitor (U0126), suggesting that LPS-stimulated cPLA2α phosphorylation and activity are mediated through an ERK-dependent mechanism. Moreover, COX-2-derived PGE2 production appeared to involve in LPS-induced VCAM-1 expression which was attenuated by pretreatment with selective COX-2 inhibitors (NS-398 and celecoxib), transfection with COX-2 siRNA, or PGE2 receptor antagonists. In addition, pretreatment with ecosapentaenoic acid (EPA), a substrate competitor of arachidonic acid (AA), also blocked LPS-induced VCAM-1 mRNA and protein expression, and THP-1 adherence. Collectively, these results suggest that LPS-induced VCAM-1 expression and adhesion of THP-1 cells are mediated through the TLR4/ERK/cPLA2α phosphorylation and COX-2 expression/PGE2 synthesis in RASFs. J. Cell. Physiol. 223: 480–491, 2010. © 2010 Wiley-Liss, Inc.
Rheumatoid arthritis (RA) is a chronic inflammatory disease manifested by persistence of inflammatory responses and a stable infiltration of lymphocytes in multiple joints. Several types of cytokines, chemokines, and adhesion molecules can be produced by RA synovial fibroblasts (RASFs) and alter the behavior of infiltrating leukocytes, leading to their inappropriate survival and retention (Carter and Wicks, 2001; Carter et al., 2002; Buckley, 2003). Adhesion molecules such as vascular adhesion molecule-1 (VCAM-1) induced by pro-inflammatory substances such as lipopolysaccharide (LPS), predominantly bind with the integrin α4β1 which is expressed by most circulating leukocytes (Elices et al., 1990). Thus, leukocytes can be recruited to sites of inflammation via interaction between VCAM-1 and α4β1 integrin which causes monocytes/macrophages accumulation in joints, these are main causes of persistent synovial inflammation in RA patients (Carter and Wicks, 2001). Accumulating evidence indicates that VCAM-1 is strongly expressed on several cell types in RA synovium, including fibroblast-like synovial lining cells (Wilkinson et al., 1993), chondrocytes (Kienzle and von Kempis, 1998), and endothelial cells (Koch et al., 1991; Wilkinson et al., 1993). Furthermore, collagen-induced joint arthritis and B cell trafficking can be reduced by the blockade of VCAM-1 (Carter et al., 2002). Therefore, VCAM-1 overexpression may be implicated in pathogenesis of RA, but the mechanisms underlying LPS-induced VCAM-1 up-regulation are still unknown in RA patients.
Toll-like receptors (TLRs) are pattern recognition receptors that are critical for the generation of both innate and adaptive immunity. It has been demonstrated that TLR4 is expressed by macrophages and fibroblasts in synovial lining and up-regulated in inflamed synovium of RA (Huang et al., 2007; Sacre et al., 2007; Ospelt et al., 2008), and detects the self matrix component such as biglycan, heparan sulfate, extra domain A of fibronectin, and fibrinogen (Cleland et al., 2003; O'Neill, 2008; Mickleborough et al., 2009). It has been established that activation of macrophages by LPS is mediated through TLR4. Inhibition of the TLR4 pathway substantially suppresses pathological features of collagen-induced joint arthritis model (Abdollahi-Roodsaz et al., 2007). In addition, persistent joint inflammation in the latter phase of streptococcal cell wall-induced arthritis is significantly inhibited in TLR4−/− mice (Abdollahi-Roodsaz et al., 2008). In addition, the activation of TLR4 in articular chondrocytes decreases matrix biosynthesis, suggesting that TLR4 may hamper the repair of cartilage in various joint diseases (Bobacz et al., 2007). However, the mechanisms involved in LPS/TLR4-mediated expression of inflammatory proteins in RASFs are still unclear.
Prostaglandins (PGs) and other arachidonic acid (AA) metabolites that are collectively designated “eicosanoids” are involved in inflammatory responses or autoimmune diseases such as allergic rhinitis, RA, and asthma (Shimizu et al., 2006). The initial step of eicosanoid biosynthesis is the release of AA from membrane phospholipids by cytosolic phospholipase A2α (cPLA2α) activation; AA is in turn converted to PGs and thromboxanes by cyclooxygenase (COX) and to leukotrienes by 5-lipoxygenase, based on the observation of these responses being blunted in cPLA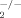 model (Sapirstein and Bonventre, 2000). Several studies have demonstrated that inhibition of AA metabolism attenuates symptoms of RA (Simopoulos, 2002; Hegen et al., 2003; Mori and Beilin, 2004), suggesting that cPLA2α plays a critical role in pathogenesis of collagen-induced joint arthritis model and inhibition of cPLA2α expression by antisense oligonucleotides may serve as an efficient treatment of inflammatory diseases (Raichel et al., 2008). AACOCF3, a selective cPLA2 inhibitor, also prevents inflammatory diseases such as allergen-induced airway hyper-responsiveness, hypoxic pulmonary vasoconstriction, and multiple sclerosis in rodent model (Malaviya et al., 2006). Therefore, the cPLA2α-induced AA cascade is harmful to the RA. cPLA2α activity has been reported to be regulated by phosphorylation at Ser505, Ser515, and Ser727 by mitogen-activated protein kinases (MAPKs), such as ERK1/2 and p38 (Pavicevic et al., 2008; Tian et al., 2008). Those MAPKs have been reported to be activated by TLR4 in RASFs. In some systems, cPLA2α activation regulates the transcriptional events that culminate in the induction of several inflammatory proteins, such as COX-2 (Sapirstein et al., 2005), inducible NO synthase (Pindado et al., 2007), and IL-6 and IL-1β (Wang et al., 2008). In RASFs, the role of LPS-stimulated cPLA2α activation implicated in RA inflammation is still unknown.
model (Sapirstein and Bonventre, 2000). Several studies have demonstrated that inhibition of AA metabolism attenuates symptoms of RA (Simopoulos, 2002; Hegen et al., 2003; Mori and Beilin, 2004), suggesting that cPLA2α plays a critical role in pathogenesis of collagen-induced joint arthritis model and inhibition of cPLA2α expression by antisense oligonucleotides may serve as an efficient treatment of inflammatory diseases (Raichel et al., 2008). AACOCF3, a selective cPLA2 inhibitor, also prevents inflammatory diseases such as allergen-induced airway hyper-responsiveness, hypoxic pulmonary vasoconstriction, and multiple sclerosis in rodent model (Malaviya et al., 2006). Therefore, the cPLA2α-induced AA cascade is harmful to the RA. cPLA2α activity has been reported to be regulated by phosphorylation at Ser505, Ser515, and Ser727 by mitogen-activated protein kinases (MAPKs), such as ERK1/2 and p38 (Pavicevic et al., 2008; Tian et al., 2008). Those MAPKs have been reported to be activated by TLR4 in RASFs. In some systems, cPLA2α activation regulates the transcriptional events that culminate in the induction of several inflammatory proteins, such as COX-2 (Sapirstein et al., 2005), inducible NO synthase (Pindado et al., 2007), and IL-6 and IL-1β (Wang et al., 2008). In RASFs, the role of LPS-stimulated cPLA2α activation implicated in RA inflammation is still unknown.
Although the expression of VCAM-1, cPLA2α, and COX-2 induced by various stimuli has been extensively investigated, it is not yet known whether cPLA2α activation and COX-2 expression are functionally coupled with VCAM-1 expression and cell adhesion in RASFs. In this study, we found that LPS-induced VCAM-1 expression and adhesion of THP-1 cells was mediated through ERK-dependent cPLA2α phosphorylation and COX-2 expression/PGE2 synthesis in these cells. These findings support a novel mechanism of LPS/TLR4 activation to regulate synovial inflammation which serves as a potential therapeutic target in the treatment of RA.
Materials and Methods
Materials
DMEM/F-12 medium, FBS, BCECF-AM, RPMI-1640, OPTI-MEM, and TRIzol were from Invitrogen (Carlsbad, CA). Hybond C membrane, enhanced chemiluminescence (ECL) Western blotting detection system and Hyperfilms were from GE Healthcare Biosciences (Buckinghamshire, England). Polyclonal antibody VCAM-1 (cat. sc-1504) was from Santa Cruz (Santa Cruz, CA). Anti-GAPDH (cat. #4699-9555) antibody was from Biogenesis (Bournemouth, UK). Anti-phospho-p42/p44 MAPK (cat. #4376) and anti-phospho-cPLA2α (cat. #2831) antibodies were from Cellular Signalling (Danver, MA). Anti-TLR4 (cat. IMG-417E) antibody was from Imgenex (San Diego, CA). Control IgG (cat. 16-4724-85) antibody was from eBioscience (San Diego, CA). U0126 and AACOCF3 were from Biomol (Plymouth Meeting, PA). Bicinchoninic acid (BCA) protein assay kit was from Pierce (Rockford, IL). cPLA2 assay kit, PGE2 EIA kit, Eicosa-5Z, 8Z, 11Z, 14Z, 17Z-pentaenoic acid (EPA; 20:5, n-3), NS-398, SC-19200, AH 6809, and GW627368X were from Cayman Chemicals (Ann Arbor, MI). Enzymes, LPS (E. coli 0111:B4), and other chemicals were from Sigma (St. Louis, MO).
Culture of synovial fibroblasts
The primary cultured human synoviocytes (HSs) was purchased from ScienCell (San Diego, CA). RA synovial tissues were obtained from patients with RA who underwent knee or hip surgery. RA patients fulfilled the criteria of the American College of Rheumatology. Informed consent was obtained from all patients, and the experimental protocol was approved by the Institutional Review Board, Chang Gung Memorial Hospital. RA synovial tissues were cut into small pieces and placed in 10 cm-dishes. These cells were grown in DMEM/F-12 containing 10% FBS and antibiotics (100 U/ml penicillin G, 100 µg/ml streptomycin, and 250 ng/ml fungizone) at 37°C in a humidified 5% CO2 atmosphere. When the cultures reached confluence, cells were treated with 0.05% (w/v) trypsin/0.53 mM EDTA for 5 min at 37°C. The cell suspension were diluted with DMEM/F-12 containing 10% FBS to a concentration of 2 × 105 cells/ml. Over 95% of the cells were fibroblasts which were characterized by an immunofluorescence staining using an antibody specific for a fibroblast protein vimentin. Culture medium was changed after 24 h and then every 3 days. Experiments were performed using cells from passages 3–8.
Adhesion assay
The human monocytic cell line THP-1 was kindly provided by Dr. Shyi-Wu Wang (Department of Physiology, Chang Gung University). THP-1 cells were labeled with 10 µM BCECF-AM at 37°C for 1 h in RPMI-1640 medium and subsequently washed by centrifugation. RASFs grew on glass coverslips were incubated with LPS for 6 h. Confluent LPS-treated RASFs were incubated with THP-1 cells (2 × 106 cells/ml) at 37°C for 1 h. Non-adherent THP-1 cells were removed and gently washed with PBS. The number of adherent THP-1 cells was counted under four fields per 200× high power using a fluorescent microscopy (Axiovert 200M, Carl Zeiss MicroImaging GmbH, Jena, Germany). Four randomly chose high-power fields were counted per well.
Preparation of cell extracts and Western blot analysis
RASFs were plated onto 12-well culture plates and made quiescent by incubation in DMEM/F-12 contain 1% FBS for 24 h. Growth-arrested cells were incubated with LPS (100 µg/ml) or PGE2 (0.3, 3, 30 µM) at 37°C for the indicated time intervals in 1% serum-containing medium. When inhibitors (U0126, AACOCF3, NS-398, SC-19200, AH 6809, GW627368X, or EPA) were used, they were added 1 h prior to the addition of agonist. The whole cell lysates were prepared and subjected to SDS–PAGE as previously described (Wu et al., 2004). Membranes were incubated overnight at 4°C with an anti-phospho-p42/p44 MAPK, anti-phospho-cPLA2α, anti-VCAM-1, anti-GAPDH, or IgG antibody used at a dilution of 1:2000 in TTBS. Membranes were washed with TTBS four times for 5 min each, incubated with a 1:1500 dilution of anti-rabbit or anti-mouse horseradish peroxidase antibody for 1 h. Following each incubation, the membranes were washed extensively with TTBS. The immunoreactive bands detected by ECL reagents were developed by Hyperfilm-ECL.
Total RNA extraction and real-time RT-PCR analysis
Total RNA was isolated from RASFs treated with LPS for the indicated time intervals in 10-cm culture dishes with TRIzol according to the protocol of the manufacturer. RNA concentration was spectrophotometrically determined at 260 nm. Real-time PCR was performed with the TaqMan gene expression assay system, using primer and probe mixes for VCAM-1 and endogenous GAPDH control genes. PCRs were performed using the 7500 real-time PCR system (Applied Biosystems, Foster City, CA). Relative gene expression was determined by the ΔΔCt method, where Ct = threshold cycle. All experiments were performed in duplicate or triplicate.
Transfection with siRNA
Human cPLA2α (OligioID: HSS108067; cat. #1299003) and negative control siRNA duplexes (cat. #12935-300) were from Invitrogen; COX-2 siRNA (target sequence: GGACUUAUGGGUAAUGUUA) was from Thermo Fisher Scientific (Pittsburgh, PA). The siRNA was transfected into RASFs using Lipofectamine RNAiMAX according to the protocol of the manufacturer (Invitrogen). Briefly, prior to transfection, the cultures were washed with PBS and replaced with 2.5 ml/well (six-well plate) of OPTI-MEM. The siRNA was transfected at 30 nM with Lipofectamine RNAiMAX (5 µl/well). The transfection reactions were changed to fresh culture media containing 10% FBS after 4 h. After 36 h transfection, the cells from some wells were harvested and determined expression of cPLA2, COX-2, and GAPDH by using Western blot. The cells from the remaining wells were then stimulated with LPS (100 µg/ml) for the indicated time intervals.
Measurement of cPLA2 activity
Cells were washed with PBS, incubated in DMEM/F-12 medium containing 1% serum for 24 h, and then treated with 100 µg/ml LPS. The cell lysates were collected to measure cPLA2 activity using a cPLA2 assay kit. To avoid the measurement of secretory PLA2 (sPLA2) and Ca2+-independent PLA2 (iPLA2), the sPLA2-specific inhibitor, thioetheramide-PC, and the iPLA2-specific inhibitor, bromoenol lactone, were added to the samples prior to the assay.
Measurement of PGE2 production
Cells were washed with PBS, incubated in DMEM/F-12 medium containing 1% serum for 24 h, and then treated with 100 µg/ml LPS. The culture supernatant was collected to measure PGE2 concentration using an EIA kit as specified by the manufacturer (Cayman Chemicals).
EPA treatment
RASFs were pretreated with the indicated concentrations of EPA from stock solutions in ethanol (EtOH, final concentration ≤0.2%), and then incubated with LPS (100 µg/ml) for 1, 4, or 6 h. Control cultures always included ethanol at the appropriate concentration, and the solvent itself had no effect on biological activity of RASFs.
Statistical analysis
Concentration–effect curves were made and EC50 values were estimated using the GraphPad Prism Program (GraphPad, San Diego, CA). Quantitative data were expressed as the mean ± SEM and analyzed with a one-way ANOVA to make comparisons with Bonferroni's test at a P < 0.05 level of significance. Error bars were omitted when they fell within the dimensions of the symbols.
Results
LPS induces VCAM-1 expression in RASFs
To determine whether LPS-induced VCAM-1 expression, RASFs were incubated with LPS (100 µg/ml) for the indicated time intervals. LPS-induced VCAM-1 expression in a time-dependent manner with a maximum within 6 h during the period of observation (Fig. 1A). There was no change in GAPDH (as an internal control) expression. To illustrate whether LPS-induced VCAM-1 protein expression occurred at transcriptional level, LPS-induced VCAM-1 mRNA expression was determined by real-time RT-PCR. As shown in Figure 1B, LPS-induced VCAM-1 mRNA expression was significantly increased with a maximum within 2 h. These results suggested that LPS-induced VCAM-1 expression through up-regulating mRNA and protein levels. To test the functional activity of VCAM-1 expressed on RASFs, as shown in Figure 1C, the extent of THP-1 adherence to RASFs incubated with LPS was significantly increased (∼5- to 6-folds) which was concentration-dependently inhibited by pretreatment with an anti-VCAM-1 antibody. Next, to investigate the involvement of TLR4 in LPS-induced VCAM-1 expression and increase THP-1 adherence, RASFs were pretreated with an anti-TLR4 antibody (1, 3, or 5 µg/ml) for 1 h and then incubated with 100 µg/ml LPS for 6 h. As shown in Figure 1C, pretreatment with anti-TLR4 antibody attenuated LPS-induced VCAM-1 expression and THP-1 adherence in a concentration-dependent manner, indicating the involvement of TLR4 in these LPS-mediated responses.

LPS-induced VCAM-1 expression and THP-1 adherence. A: Time dependence of VCAM-1 protein expression, RASFs were incubated with 100 µg/ml of LPS for the indicated time intervals. The expression of VCAM-1 was determined by Western blot using an anti-VCAM-1 or IgG Ab. B: RASFs were incubated with 100 µg/ml of LPS for the indicated time intervals. Total RNA was isolated and real-time quantitative PCR for VCAM-1 and GAPDH was performed. Data were analyzed by the comparative cycle threshold method and expressed as the fold relative to those of non-stimulated cells. C: RASFs were pretreated without or with an anti-TLR4 Ab (5 µg/ml) for 1 h and then challenged with 100 µg/ml of LPS for 6 h. Whole cell lysates were analyzed by Western blot using an anti-VCAM-1 Ab. One part of LPS-challenged RASFs was pretreated with an anti-VCAM-1 (1, 3, or 5 µg/ml), anti-TLR4 (1, 3, or 5 µg/ml), or IgG (5 µg/ml) Ab for 1 h prior to performing adhesion assay. THP-1 cells labeled with BCECF-AM (10 µM) were added to RASFs, and then the THP-1 cells adherence was measured. Data are expressed as mean ± SEM of three independent experiments from three different RASFs preparations. Significant differences between the compared groups are indicated: *P < 0.05; #P < 0.01, as compared with the cells exposed to vehicle alone (A,B) or LPS alone (C).
LPS induces VCAM-1 expression via cPLA2α activation
It has been shown that LPS could activate cPLA2α leading to the expression of several target proteins in various cell types (Qi and Shelhamer, 2005; Kikawada et al., 2007; Pindado et al., 2007). Thus, to determine whether expression of VCAM-1 was regulated by cPLA2α activation, RASFs were pretreated with an inhibitor of cPLA2 activity (AACOCF3) for 1 h and then exposed to LPS (100 µg/ml) for 6 h. As shown in Figure 2A, LPS-induced VCAM-1 expression was significantly inhibited by pretreatment with AACOCF3. Next, we determined whether LPS-stimulated cPLA2α activity in RASFs. cPLA2α phosphorylation at Ser505 residue has been shown to be required for its activation. Thus, the cPLA2α phosphorylation at Ser505 was determined by Western blot using an anti-phospho-cPLA2α antibody. The data showed that LPS-stimulated cPLA2α phosphorylation in a time-dependent manner with a maximum within 60 min and sustained for up to 180 min (Fig. 2B). To ensure the role of cPLA2α activation in LPS-induced VCAM-1 expression, RASFs were transfected with cPLA2α siRNA. As shown in Figure 2C, transfection of RASFs with cPLA2α siRNA (30 nM) for 36 h significantly knocked down the expression of cPLA2α protein and attenuated the LPS-induced VCAM-1 expression, but had no effect on housekeeping GAPDH protein expression (Fig. 2C). These results suggested that cPLA2α activation is involved in LPS-induced VCAM-1 expression in RASFs.

Activation of cPLA2α is essential for LPS-induced VCAM-1 expression. A: RASFs were pretreated with AACOCF3 for 1 h and then challenged with LPS for 6 h. The cell lysates were determined by Western blot using an anti-VCAM-1 Ab. B: Time dependence of cPLA2α protein phosphorylation, RASFs were incubated with 100 µg/ml of LPS for the indicated time intervals. The phosphorylation of cPLA2α was determined by Western blot using an anti-phospho-cPLA2α Ab. Membranes were stripped and re-probed with an anti-cPLA2, anti-GAPDH, or IgG Ab. C: The cells were transfected with a cPLA2α siRNA (30 nM) for 36 h and then incubated with LPS for 6 h. The cell lysates were assayed by Western blot using an anti-cPLA2, anti-phospho-cPLA2α, or anti-VCAM-1 Ab. Membranes were stripped and re-probed with an anti-GAPDH Ab as an internal control. Data are expressed as mean ± SEM of three independent experiments from three different RASFs preparations. #P < 0.01, as compared with the cells exposed to LPS alone.
LPS-induced VCAM-1 expression is mediated through ERK1/2-dependent cPLA2α activation
The MAPK pathway is one of the major downstream pathways in TLR4 signaling (Qi and Shelhamer, 2005). Moreover, it has been shown that the cPLA2α activity is regulated by MAPKs, including p38 MAPK and ERK1/2, resulting in phosphorylation of cPLA2α at Ser505 (Qi and Shelhamer, 2005; Kikawada et al., 2007; Nito et al., 2008; Pavicevic et al., 2008). Therefore, we next investigated whether the LPS-induced VCAM-1 expression was mediated through MAPK-dependent cPLA2α activation. Data in Figure 3A show that LPS-stimulated phosphorylation of cPLA2α was attenuated by pretreatment with an inhibitor of MEK1/2 (U0126), but not by a p38 MAPK inhibitor (SB202190), suggesting that LPS-stimulated phosphorylation of cPLA2α was mediated through ERK1/2 pathway in RASFs. Moreover, LPS stimulated a time-dependent phosphorylation of ERK1/2 which was attenuated by pretreatment with U0126 in a concentration-dependent manner (Fig. 3B). Similarly, LPS-increased cPLA2α activity (Fig. 3C) and VCAM-1 expression (Fig. 3D) were markedly attenuated by pretreatment with U0126. Thus, LPS-induced VCAM-1 expression was mediated through an ERK-dependent cPLA2α phosphorylation in RASFs. Moreover, pretreatment with U0126 or AACOCF3 also attenuated LPS-induced VCAM-1 mRNA expression (Fig. 3E) and THP-1 cell adherence (Fig. 3F). These results suggested that ERK1/2-dependent cPLA2α activation signaling is essential for LPS-induced VCAM-1 expression and THP-1 cells adherence to RASFs. To further ensure that cPLA2α was involved in LPS-induced THP-1 cell adherence, as shown in Figure 3F, transfection with cPLA2α siRNA (30 nM) attenuated THP-1 cell adherence on RASFs challenged with LPS.

LPS-induced VCAM-1 expression and THP-1 adherence mediated through ERK1/2-dependent cPLA2α activity. A: RASFs were pretreated with U0126 or SB202190 for 1 h, and then stimulated with 100 µg/ml of LPS for 60 min. The levels of cPLA2α phosphorylation were measured by Western blot using an anti-phospho-cPLA2α Ab. B: RASFs were pretreated with U0126 for 1 h, and then incubated with 100 µg/ml of LPS for 60 min. The cell lysates were analyzed by Western blotting using an anti-phospho-p42/p44 MAPK or IgG Ab. C: RASFs were pretreated with U0126 for 1 h and then incubated with LPS for the indicated time. cPLA2α activity was measured using arachidonyl thioetheramide-PC as a synthetic substrate. D: RASFs were pretreated with U0126 for 1 h and then incubated with 100 µg/ml of LPS for 6 h. The cell lysates were analyzed by Western blotting using an anti-VCAM-1Ab. E: RASFs were pretreated with 1 µM U0126 or 1 µM AACOCF3 for 1 h and then stimulated with 100 µg/ml of LPS for 4 h. Total RNA was isolated and real-time quantitative PCR for VCAM-1 and GAPDH was performed. Data were analyzed by the comparative cycle threshold method and expressed as the fold relative to those of non-stimulated control. F: RASFs were pretreated with 1 µM U0126, 1 µM AACOCF3 or transfected with siRNA (scramble or cPLA2) and then challenged with LPS for 6 h. THP-1 cells labeled with BCECF-AM (10 µM) were added to RASFs, and then the THP-1 cells adherence was measured. Data are expressed as mean ± SEM of three independent experiments from three different RASFs preparations. *P < 0.05; #P < 0.01, as compared with the cells exposed to LPS alone.
Involvement of PGE2 in LPS-induced VCAM-1 expression
The data in Figure 3 indicate that cPLA2α plays a pivotal role in LPS-induced VCAM-1 expression and THP-1 adherence. In mammalian cells, AA, a principal product of cPLA2α, has been known to be catalyzed by COX to form PGs. To dissect out the downstream component of cPLA2α via TLR4 signaling associated with VCAM-1 expression, the selective COX-2-inhibitors were used. As shown in Figure 4A, pretreatment with two selective COX-2 inhibitors, NS-398, and celecoxib (CLC), respectively, significantly inhibited LPS-induced VCAM-1 expression. These findings prompted us to postulate that PGE2, a principal metabolic product generalized by COX-2, could involve in VCAM-1 expression induced by LPS in RASFs. Thus, the PGE2 receptor antagonists SC-19200, AH 6809, and GW627368X were used. As shown in Figure 4B, pretreatment with respective PGE2 receptor antagonist attenuated LPS-induced VCAM-1 expression, suggesting that PGE2 is involved in LPS-induced VCAM-1 expression. Thus, we further tested whether LPS could regulate COX-2 expression and PGE2 synthesis in RASFs. The cells were incubated with LPS for the indicated time intervals. LPS-induced COX-2 expression and PGE2 synthesis in a time-dependent manner in RASFs (Fig. 4C, upper part, D). To further ensure that COX-2 was involved in VCAM-1 expression, as shown in Figure 4C (middle part), transfection with COX-2 siRNA (30 nM) knocked down the expression of COX-2 protein and attenuated LPS-induced VCAM-1 expression. These data suggested a functional link between COX-2 activity and VCAM-1 expression. The data obtained from transfection with cPLA2α and COX-2 siRNA indicate that both cPLA2α and COX-2 are involved in LPS-induced VCAM-1 expression. Therefore, we further ensure that cPLA2α/COX-2 pathway is involved in LPS-induced VCAM-1 expression. As shown in Figure 4C (lower part), cotransfection with a cPLA2α (30 nM) and COX-2 (30 nM) siRNA for 36 h and significantly attenuated LPS-induced VCAM-1 expression. Next, we examined the role of ERK-dependent cPLA2α/COX-2 signaling in LPS-induced PGE2 release, RASFs were pretreated with U0126 for 1 h or transfected with cPLA2α or COX-2 siRNA. As shown in Figure 4E, pretreatment with U0126 or transfection with cPLA2α and COX-2 siRNA significantly inhibited LPS-induced PGE2 production. These results suggested that LPS-induced PGE2 formation is mediated by an ERK/cPLA2α/COX-2 signaling pathway. To ensure that PGE2 played an important role in LPS-induced VCAM-1 expression, RASFs were incubated with various concentrations of PGE2 for the indicated time intervals. As shown in Figure 4F, PGE2-induced VCAM-1 expression in a time- and concentration-dependent manner. Moreover, pretreatment with NS-398, CLC, AACOCF3, and U0126 had suppressive effects on LPS-induced VCAM-1 protein expression. The attenuation of VCAM-1 protein expression by these inhibitors was restored by exogenous addition of (1 nM) PGE2 (data not shown). These results indicated that cPLA2α/COX-2-mediated PGE2 synthesis cooperatively induced VCAM-1 expression induced by LPS in RASFs.

COX-2/PGE2 is involved in LPS-induced VCAM-1 expression. RASFs were pretreated with (A) selective COX-2 inhibitors (NS-398 and celecoxib) or (B) PGE2 receptor antagonists (SC-19200, AH6809, and GW627368X) for 1 h and then incubated with 100 µg/ml of LPS for 6 h. The cell lysates were analyzed by Western blotting using an anti-VCAM-1 Ab. C: Time dependence of COX-2 protein expression, RASFs were incubated with 100 µg/ml of LPS for the indicated time intervals. The cell lysates were analyzed by Western blotting using an anti-COX-2 or IgG Ab. C, middle part: The cells were transfected with a COX-2 siRNA (30 nM) for 36 h and incubated with LPS for 6 h. C, lower part: The cells were cotransfected with a cPLA2αα and COX-2 siRNA for 36 h and incubated with LPS for 6 h. The cell lysates were assayed by Western blot using an anti-VCAM-1 or anti-COX-2 Ab. Membranes were stripped and re-probed with an anti-GAPDH Ab as an internal control. D: Time dependence of PGE2 synthesis, RASFs were incubated with 100 µg/ml of LPS for the indicated time intervals. E: The cells were pretreated with (1 µM) U0126 for 1 h, or transfected with cPLA2α (30 nM) and COX-2 (30 nM) siRNA, and then incubated with 100 µg/ml of LPS for 6 h. D and E: The media were saved and detected by a PGE2 ELISA kit. F: Time dependence of VCAM-1 protein expression, RASFs were incubated with PGE2 for the indicated time intervals. The cell lysates were analyzed by Western blotting using an anti-VCAM-1 Ab. Data are expressed as mean ± SEM of three independent experiments from three different RASFs preparations. *P < 0.05; #P < 0.01, as compared with the cells exposed to vehicle alone (D) or LPS alone (E).
The ERK/cPLA2α/COX-2/PGE2 pathway is involved in LPS-induced VCAM-1 expression in normal human synoviocytes
Next, we also determined the effect of LPS on expression of VCAM-1 by primary cultured HSs. We found that treatment of RASFs or HSs with LPS-induced VCAM-1 expression in a time-dependent manner (Fig. 5A). Next, to demonstrate whether ERK/cPLA2/COX-2/PGE2 pathway was also involved in the LPS-induced VCAM-1 expression in HSs, as shown in Figure 5B, pretreatment with U0126, AACOCF3, celecoxib, SC-19200, AH 6809, or GW627368X attenuated LPS-induced VCAM-1 expression, suggesting that LPS-induced VCAM-1 expression is mediated through an ERK/cPLA2/COX-2/PGE2 pathway.

LPS-induced VCAM-1 expression through ERK/cPLA2/COX-2/PGE2 pathway in normal human synoviocytes. A: Time dependence of VCAM-1 protein expression, RASFs and HSs were incubated with 100 µg/ml of LPS for the indicated time intervals. The expression of VCAM-1 was determined by Western blot using an anti-VCAM-1. Membranes were stripped and re-probed with an anti-GAPDH Ab as an internal control. B: RASFs were pretreated with U0126, AACOCF3 selective COX-2 inhibitors (celecoxib) or PGE2 receptor antagonists (SC-19200, AH6809, and GW627368X) for 1 h and then incubated with 100 µg/ml of LPS for 6 h. The cell lysates were analyzed by Western blotting using an anti-VCAM-1 Ab. Membranes were stripped and re-probed with an anti-GAPDH Ab as an internal control. Data are expressed as mean ± SEM of three independent experiments. *P < 0.05; #P < 0.01, as compared with the cells exposed to LPS alone.
EPA blocks LPS-induced VCAM-1 expression by diminishing biosynthesis of PGE2
According to the findings mentioned above, we suggested that LPS-induced VCAM-1 expression is modulated by activation of the cPLA2α/COX-2/PGE2 signaling pathway. Therefore, we ascertained that an AA competitor, EPA, could competitively block LPS-induced VCAM-1 expression. As shown in Figure 6A, LPS-induced VCAM-1 protein and mRNA expression were reduced by pretreatment with EPA in RASFs (Fig. 6B). Moreover, pretreatment with EPA also attenuated THP-1 cells adherence to RASFs challenged with LPS (Fig. 6C). However, EPA (20 µM) failed to attenuate the LPS stimulated the activation of ERK1/2 and cPLA2α (Fig. 6D). To examine whether the effect of EPA on LPS-induced VCAM-1 expression due to the attenuation of PGE2 synthesis, as shown in Figure 6E, pretreatment with EPA significantly inhibited LPS-induced PGE2 production in RASFs. These results implied that EPA attenuated LPS-induced VCAM-1 expression and THP-1 cells adhesion due to attenuating PGE2 synthesis in these cells.

EPA attenuated LPS-induced VCAM-1 expression and THP-1 adherence through reduction of PGE2 synthesis. A: RASFs were pretreated with EPA for 1 h and then challenged with 100 µg/ml of LPS for 6 h. The cell lysates were analyzed by Western blotting using an anti-VCAM-1 Ab. B: RASFs were pretreated with (20 µM) EPA for 1 h and then incubated with LPS for 4 h. Total RNA was isolated and real-time quantitative PCR for VCAM-1 and GAPDH was performed. Data were analyzed by the comparative cycle threshold method and expressed as the fold relative to those of the non-stimulated cells. C: RASFs were pretreated with (20 µM) EPA for 1 h and then incubated with LPS for 6 h. THP-1 cells labeled with BCECF-AM (10 µM) were added to RASFs, and then the THP-1 cells adherence was measured. D: The cells were pretreated with EPA for 1 h and then incubated with 100 µg/ml LPS for 60 min. The cell lysates were analyzed by Western blotting using an anti-phospho-p42/p44 MAPK, anti-phospho-cPLA2α, or anti-cPLA2 Ab. Membranes were stripped and re-probed with an anti-GAPDH Ab as an internal control. E: RASFs were pretreated with (20 µM) EPA for 1 h and then challenged with 100 µg/ml of LPS for 6 h. The media were saved and detected by a PGE2 ELISA kit. Data are expressed as mean ± SEM of three independent experiments from three different RASFs preparations. *P < 0.05; #P < 0.01, as compared with the cells exposed to LPS alone. EtOH, ethanol.
Discussion
In RA, VCAM-1 plays a key role in monocytes/macrophages accumulation in joints and involves in promoting survival of T and B lymphocytes to cause persistent inflammation. Several lines of evidence have shown that LPS can induce VCAM-1 expression in various cell types. Moreover, TLR4 is expressed at higher levels in RA synovial tissue than in normal synovial tissue or that from patients with osteoarthritis (Ospelt et al., 2008). The activation of TLR4 pathway has been shown to be implicated in the pathogenesis of RA (Abdollahi-Roodsaz et al., 2007, 2008; O'Neill, 2008; Ospelt et al., 2008). Therefore, the aims of this study clarified how LPS/TLR4 pathway regulated VCAM-1 expression in RA. Herein, we demonstrated that LPS-induced VCAM-1 expression and adhesion of THP-1 cells was mediated through an ERK-dependent cPLA2α/COX-2/PGE2 signaling pathway in RASFs. Moreover, EPA could decrease LPS-induced VCAM-1 expression and adhesion of THP-1 cells through reducing production of PGE2.
In this study, LPS induced the expression of VCAM-1 in protein and mRNA levels on RASFs (Fig. 1A,B). The role of VCAM-1 induction has been shown to be involved in the pathogenesis of inflammatory joint diseases, such as leukocyte adhesion to activated synovial fibroblasts and retention of infiltrating leukocytes within extravascular compartments (Carter and Wicks, 2001; Carter et al., 2002; Buckley, 2003). LPS has been shown to contribute to VCAM-1 expression in human tracheal smooth muscle cells (Lin et al., 2007). A recent study has clearly shown that LPS induces IL-1β secretion through the TLR4-signaling pathway in articular chondrocytes (Abdollahi-Roodsaz et al., 2007). In our study, we observed that LPS-induced VCAM-1 expression was inhibited by pretreatment with an anti-TLR4 antibody (Fig. 1C). These results clearly suggested that TLR4 is the central signaling receptor for LPS in RASFs. Moreover, we also observed that TLR4 was involved in THP-1 adherence to RASFs through VCAM-1 overexpression induced by LPS. This notion was supported by our results indicating that pretreatment with either VCAM-1 or TLR4 neutralizing antibody can decrease THP-1 adherence to the RASFs challenged with LPS. These data suggest that LPS/TLR4 enhances the adhesion of THP-1 cells to RASFs through VCAM-1 expression.
It has been shown that cPLA2α preferentially cleaves phospholipids at the sn-2 position to release AA. Therefore, cPLA2α is considered as the key enzyme responsible for receptor-mediated AA release and subsequent cellular responses (Kramer and Sharp, 1997). Several reports have shown that expression of various inflammatory proteins can be regulated by cPLA2α signaling pathway (Qi and Shelhamer, 2005; Kikawada et al., 2007; Pindado et al., 2007; Nito et al., 2008). In present study, we also demonstrated that LPS-induced VCAM-1 expression was mediated through cPLA2α activation in RASFs. This hypothesis was supported by results showing that pretreatment with a selective cPLA2 inhibitor (AACOCF3) or transfection with cPLA2α siRNA attenuated LPS-stimulated cPLA2α phosphorylation and subsequent VCAM-1 expression in RASFs, consistent with that cPLA2α-driven signaling cascade involved in iNOS expression (Pindado et al., 2007). It is worth noting that pretreatment with U0126 displayed stronger ability to reduce LPS-induced VCAM-1 expression than that of transfection with cPLA2α siRNA, suggesting that ERK1/2 may also mediate through additional pathways to regulate LPS-induced VCAM-1 expression.
Cytokines, chemokines, and TLRs ligand can aggravate symptoms of RA through activation of MAPKs. MAPKs are a family of serine/theronine kinases that transduce extracellular signals to the nucleus (Kramer and Sharp, 1997). Three groups of MAPKs that differ in their substrate specificity are recognized in mammalian cells: ERK1/2, JNK, and p38 MAPK. As we know, cPLA2α activity is tightly regulated by a low concentration of intracellular Ca2+, and is mediated through MAPKs and protein kinase C (Kikawada et al., 2007; Pindado et al., 2007; Nito et al., 2008). The activation of cPLA2α requires phosphorylation at multiple sites which can be regulated by p38 MAPK and ERK1/2 activation. Previous studies have indicated that ERK1/2 or p38 MAPK phosphorylates cPLA2α at Ser505 and then promotes cPLA2α activity in several cell types (Qi and Shelhamer, 2005; Kikawada et al., 2007; Pavicevic et al., 2008; Lindner et al., 2009). Our data provide evidence that LPS triggered cPLA2α phosphorylation at Ser505 through an ERK-dependent pathway, but not p38 MAPK (Figs. 2B and 3A), and thus enhanced its activity (Fig. 3C). These results were consistent with previous reports indicating that ERK1/2-dependent cPLA2α phosphorylation in response to TLR2 stimulation has also been found in mast cells (Kikawada et al., 2007).
Our data showed that LPS-induced COX-2 expression occurred earlier than that of VCAM-1 expression (Figs. 1A and 4C, upper part). We also found that pretreatment with the COX-2 inhibitors (celecoxib or NS-398) and transfection with COX-2 siRNA or cotransfection with cPLA2α and COX-2 could attenuate LPS-induced VCAM-1 expression in RASFs (Fig. 4A,C). Our data also demonstrated that LPS could enhance PGE2 production (Fig. 4D), consistent with that of COX-2 protein expression (Fig. 4C). Moreover, LPS-induced PGE2 synthesis was mediated through an ERK/cPLA2α/COX-2 pathway (Fig. 4E). The effects of PGE2 are coupled to four different receptor subtypes, including EP1, EP2, EP3, and EP4. Among them, EP1 and EP4 have been described to be highly expressed in RASFs (Mathieu et al., 2008). In this study, we demonstrated that pretreatment with EP-1 and EP-4 antagonists can significantly decrease LPS-induced VCAM-1 expression (Fig. 4B). Moreover, incubation with PGE2 alone could induce VCAM-1 expression in RASFs (Fig. 4F). These data suggested that the PGE2 generation by LPS through an ERK/cPLA2α/COX-2 cascade binds to its receptors such as EP1 or EP4 to induce VCAM-1 expression in RASFs, consistent with a report indicating that the COX-2-derived PGE2 signaling pathway is involved in LPS-induced IL-6 production in J774.2 macrophages (Chen and Lin, 1999). It is interesting to note here that treatment with COX-2 inhibitors (Fig. 4A) or PGE2 receptor antagonists (Fig. 4B) only partially block the LPS-induced VCAM-1 expression by RASFs. In addition, the cells were challenged with PGE2 (0.3 or 3 µM) alone failed to increase VCAM-1 expression (Fig. 4F), suggesting that the other AA metabolites may contribute to the responses to LPS in RASFs. In this study, we demonstrated that ERK/cPLA2/COX-2/PGE2 pathway played an important role in LPS-induced VCAM-1 expression in RASFs. According to our results, the generation of PGE2 was induced within 2 h after LPS treatment. When we used PGE2 to directly induce VCAM-1 expression with a maximal response within 30 min in RASFs, since the kinetics of LPS-induced PGE2 production through COX-2 expression may be escaped.
Animal experiments and clinical intervention studies have indicated that EPA exerts anti-inflammatory properties and might be useful in the management of RA (Simopoulos, 2002; Cleland et al., 2003; Mori and Beilin, 2004). The effects of EPA are attributed to its ability to compete with AA for enzymatic metabolism leading to less amount of inflammatory mediator release. In this study, we found that LPS-induced VCAM-1 expression via cPLA2α/COX-2-dependent PGE2 production. Recently, several studies have shown that EPA attenuates IL-1-induced stress and secretion of PGE2 (Song et al., 2003; Song and Horrobin, 2004). EPA has also been shown to reduce PLA2 expression induced by IL-1β (Song et al., 2007). In this study, we confirmed that EPA could diminish LPS-induced VCAM-1 expression and THP-1 adherence through reduction of PGE2 synthesis (Fig. 6A,C) and THP-1 adherence (Fig. 6C), but had no effect on LPS-stimulated phosphorylation of ERK1/2 and cPLA2α (Fig. 6D), consistent with the results indicating that pretreatment of human neutrophils with EPA has no effect on AA-induced phosphorylation of ERK1/2 (Moghaddami et al., 2007). In contrast, several studies have reported that pretreatment with EPA could attenuate the phosphorylation of ERK1/2 after LPS stimulation in various cell types (Lo et al., 2000; Babcock et al., 2003; Moon et al., 2007). The different results of EPA on phosphorylation of ERK1/2 may be due to cell type-specific and different experimental conditions. It is worth noting that pretreatment with EPA displayed stronger ability to reduce LPS-induced VCAM-1 expression than those of transfection with cPLA2α and COX-2 siRNA, implying that alternative signal pathways independent of the cPLA2α/COX-2 may be also involved in the induction of VCAM-1.
In this study, we demonstrated that LPS-induced VCAM-1 expression was mediated through activation of ERK/cPLA2α/COX-2 signaling pathway in RASFs. Newly synthesized PGE2 might trigger PGE2 receptors and enhance the LPS-induced VCAM-1 expression through a positive feedback control in RA. Based on the observations from literatures and our findings, Figure 7 depicts a model for molecular mechanisms underlying LPS-induced VCAM-1 expression and leukocytes adhesion in RASFs. These findings suggest that the increased expression of VCAM-1 expression is mediated through TLR4/ERK-dependent activation of cPLA2α, COX-2 expression, and PGE2 synthesis. Thus, better understanding the mechanisms underlying LPS-induced cPLA2α/COX-2/PGE2/VCAM-1 expression would be beneficial for the therapies of RA.

Schematic representation of signaling pathways involved in LPS-induced VCAM-1 expression in RASFs. LPS binding to its receptors (TLR4) results in phosphorylation of cPLA2α through an ERK1/2-dependent pathway. The functional coupling cPLA2α activity and COX-2 expression leads to PGE2 release which results in activation of the EP1 or EP4 receptors and then enhances VCAM-1 expression and cells adhesion.
Acknowledgements
We thank Dr. Shyi-Wu Wang providing the human monocytic cell line THP-1 (Department of Physiology, Chang Gung University) and Mr. Li-Der Hsiao for his assistance in preparation of this manuscript in this study.

