

Extracellular acidification enhances DNA binding activity of MafG-FosB heterodimer
Abstract
Cells are quite sensitive to a change of the extracellular pH and respond to it through detection of the H+/HCO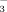 level in extracellular fluid. However, little is known about molecular details induced by acidosis, such as intracellular pathways and gene expression. Here we describe properties of gene expression, protein interaction, and DNA binding activity of basic region leucine zipper (bZIP) transcription factor Maf and FosB during extracellular acidification. When cells were incubated with low pH medium, the expressions of small Maf proteins (MafG, MafK, and MafF) and FosB were clearly increased in an extracellular pH-dependent manner and expressed transiently with a peak after 1–2 h after stimulation. Immunofluorescence and protein binding studies indicated that MafG was partially co-localized with FosB in the nucleus and MafG can form heterodimers with FosB at extracellular pH 7.40. Moreover, we found that MafG–FosB complexes are able to bind to AP-1 consensus sequence, TGACTCA. To investigate whether extracellular acidification influences to dimerization and DNA binding activity of MafG and FosB, extracellular pH of cultured cells was decreased from 7.40 to 6.80. The decrease in extracellular pH led to enhanced dimerization of MafG with FosB leading to augmentation of the DNA binding activity of the heterodimer to AP-1 consensus sequence. Moreover, extracellular acidification induces mRNA expression of matrix metalloproteinase-1, one of the genes that are regulated by AP-1. These results suggest that MafG–FosB complexes are involved in transcriptional regulation in response to extracellular acidification. © 2005 Wiley-Liss, Inc.
level in extracellular fluid. However, little is known about molecular details induced by acidosis, such as intracellular pathways and gene expression. Here we describe properties of gene expression, protein interaction, and DNA binding activity of basic region leucine zipper (bZIP) transcription factor Maf and FosB during extracellular acidification. When cells were incubated with low pH medium, the expressions of small Maf proteins (MafG, MafK, and MafF) and FosB were clearly increased in an extracellular pH-dependent manner and expressed transiently with a peak after 1–2 h after stimulation. Immunofluorescence and protein binding studies indicated that MafG was partially co-localized with FosB in the nucleus and MafG can form heterodimers with FosB at extracellular pH 7.40. Moreover, we found that MafG–FosB complexes are able to bind to AP-1 consensus sequence, TGACTCA. To investigate whether extracellular acidification influences to dimerization and DNA binding activity of MafG and FosB, extracellular pH of cultured cells was decreased from 7.40 to 6.80. The decrease in extracellular pH led to enhanced dimerization of MafG with FosB leading to augmentation of the DNA binding activity of the heterodimer to AP-1 consensus sequence. Moreover, extracellular acidification induces mRNA expression of matrix metalloproteinase-1, one of the genes that are regulated by AP-1. These results suggest that MafG–FosB complexes are involved in transcriptional regulation in response to extracellular acidification. © 2005 Wiley-Liss, Inc.
The pH of the arterial plasma is normally 7.4 and that of the cerebrospinal fluid (CSF) and venous plasma are slightly lower. Extracellular pH is physiologically maintained around pH 7.3–7.4, mainly through regulation of respiration and renal acid extrusion. Extracellular acidification has been implicated in the pathogenesis of several abnormal conditions and diseases including strenuous exercise, renal insufficiency, and diabetes mellitus. In the respiratory system, acidification in the cerebrospinal fluid and brain interstitial fluid is detected with neurons distributed over the ventral surface in the medulla oblongata, and induces a hyperpneic or tachypneic response.
The recent findings have advanced our understanding of molecular changes in cells exposed to low extracellular pH. For example, an increase of the H+ concentration in the CSF and brain interstitial fluid by hypercapnic stimulation (inhalation of air containing 7% CO2 for 5 min) causes expression of several basic region leucine zipper (bZIP) transcription factors, such as c-Fos and c-Jun proteins, in specific nuclei in the central nervous system (Sato et al., 1992; Miura et al., 1994a; Haxhiu et al., 1996; Teppema et al., 1997; Miura et al., 1998). c-Jun immunoreactive neurons are distributed in the respiration-related motor nuclei of the medulla oblongata and spinal cord, and in the central chemoreceptive area of the ventral surface of the medulla oblongata after hypercapnic stimulation. This observation was also confirmed by the fact that an increase in the concentration of extracellular H+ of cultured PC 12 cells induced c-jun mRNA (Shimokawa et al., 1998). The H+-induced c-jun mRNA expression was inhibited with Ca2+/calmodulin inhibitor trifluoperazine, indicating that the expression of c-jun mRNA is mediated partly by this system. These studies may clarify the mechanisms of our findings that the hyperventilatory response to the CO2 inhalation is abolished when the intercellular Ca2+ chelator BAPTA-AM (1, 2-bis [2-amino-4-fluorophenoxy] ethane-N, N, N′, N-tetraacetic acid tetraacetoxymethyl ester) is applied to the ventral surface of the medulla oblongata (Kanazawa et al., 1998). On the other hand, Kuo et al. (1998) reported that protein kinase Cα (PKCα) is a possible mediator in H+-induced c-Fos expression, and that PKCα may be activated by Ca2+. Activated PKCα could lead to an increase in the phosphorylation of Raf-1 kinase, which in turn activates mitogen-activated protein (MAP) kinases to stimulate expression of c-fos mRNA. Taken together, these reports suggest that an increase in concentration of extracellular H+ induces c-Jun/Fos expression through Ca2+/calmodulin and MAP kinase pathways.
In a recent study, we found that hypercapnic stimulation induces gene expression of MafG in the medulla oblongata of rat brain (Shimokawa et al., 2000). MafG is a member of small Maf protein family that consists of MafG, MafK, and MafF. Maf proteins regulate expressions of various genes as a nuclear transcription factor through dimerization with other bZIP proteins, e.g., p45, Fos, and Jun (Kataoka et al., 1994). Gene expression of MafG-2, a novel splice variant of MafG, is also induced when extracellular pH is shifted gradually from 7.40 to 7.20 (Shimokawa et al., 2001). We proposed that MafG and MafG-2 may be involved in signal transduction of extracellular acidification.
The medulla oblongata is also known to be the site of the central baroreceptive neurons, which express specific bZIP transcription factors after baroreceptor stimulation (Chan and Sawchenko, 1994; Miura et al., 1994b; Okada and Miura, 1997). Recently, we found that the number of MafG-immunoreactive neurons were increased significantly in cardiovascular control sites after stimulation of arterial baroreceptors by a blood pressure increase due to the pressor agent phenylephrine (Kumaki et al., unpublished data). The temporal expression pattern of MafG was very similar to that of FosB. These results suggest that MafG may play critical roles as an immediate early gene in cooperation with FosB in the signal transduction of cardiovascular regulation mediated by baroreceptive signals in the medulla oblongata. On the other hand, participation of FosB on extracellular acidification has yet to be studied.
Here we extend our previous work regarding the intracellular events following extracellular acidification. We show that extracellular acidification stimulates the expression of MafG and FosB. We also show that MafG heterodimerizes with FosB leading to the increased activation of DNA binding activity of the MafG-FosB dimer after extracellular acidification. These results suggest that MafG plays critical roles in transcriptional regulation in response to extracellular acidosis together with the Fos family proteins.
MATERIALS AND METHODS
Reagents and antibodies
Expression vector of protein in mammalian cells pcDNA3.0 was obtained from Invitrogen (Carlsbad, CA). T4 polynucleotide kinase, T4 DNA ligase and restriction endonucleases were purchased from New England Biolabs (Beverly, MA). Dulbecco's modified Eagle's medium (DMEM), RPMI 1640 medium, horse serum, fetal calf serum (FCS), and antibiotics (penicillin and streptomycin) were purchased from Invitrogen. Rabbit polyclonal anti-FosB (102), mouse monoclonal anti-FosB (C-11), goat polyclonal anti-MafG/F/K (C-18), and mouse monoclonal anti-FLAG (M2) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA) and Sigma (St. Louis, MO), respectively. Horseradish peroxidase (HRP)-conjugated donkey anti-rabbit IgG, sheep anti-mouse IgG, and donkey anti-goat IgG were purchased from Amersham Biosciences (Buckinghamshire, UK) and Santa Cruz Biotechnology, respectively. Alexa Fluor 488-conjugated goat anti-rabbit IgG and Alexa Fluor 568-conjugated goat anti-mouse IgG for immunofluorescence were obtained from Molecular Probes (Eugene, OR).
Plasmids
A cDNA clone containing the full-length of the rat FosB (GenBank accession no. XM218419) coding regions was cloned by PCR (forward: 5′-ATGTTTCAAGCTTTTCCCGG-3′; reverse: 5′-TTACAGAGCAAGAAGGGAGG-3′). PCR fragments were inserted into Eco RI-Xho I sites of pcDNA3.0 vector. A cDNA clone of rat MafG (GenBank accession no. AB026487) was generated by PCR using following primers: forward: 5′-ATGACGACCCCCAATAAAGG-3′; reverse: 5′-CTATGACCGAGCATCTGTC-3′. The PCR-generated fragments were inserted into Eco RI-Xho I sites of pcDNA3.0 vector. The FLAG tag (Sigma) was inserted at into Hind III-Eco RI site at the NH2 terminus of MafG.
Cell culture, acidification, and transfections
All cultured cells were provided by the RIKEN Cell Bank (Tsukuba, Japan) and kept in a humid atmosphere with 5% CO2 at 37°C. Chinese hamster ovary (CHO)-K1 cells were cultured in F12 medium supplemented with 10% FCS and antibiotics (100 U/ml penicillin and 100 μg/ml streptomycin). Human embryonic kidney (HEK) 293 cells were grown in DMEM supplemented with 10% FCS and antibiotics. Rat pheochromocytoma PC12 cells were maintained RPMI 1640 medium supplemented with 10% heat-inactivated horse serum, 5% FCS, and antibiotics. Since all cells are weakly adherent, they were plated on poly-D-lysine coated dishes (Biocoat, Becton Dickinson, Bedford, MA).
To extinguish expression of FosB and MafG that might have been present before decrease in extracellular pH, cells at 70%–80% confluence were made quiescent by incubation in a suitable culture medium (pH 7.40, 37°C) for 120 min. The culture medium was replaced with fresh medium (37°C) adjusted to pH 7.40, 7.20, 7.0, 6.80, and 6.60 with a pH meter and probe (ISFET, Beckman, Fullerton, CA), and the cells were incubated at 37°C for 0.5, 1, 2, 4, 8, or 12 h.
CHO-K1 cells were transfected 24 h following plating with 1.5 μg of expression plasmid for FosB and MafG using Lipofectamine 2000 transfection reagent (Invitrogen) as described in the manufacturer's instructions.
Cell lysis, immunoprecipitation, SDS–PAGE, and Western blot
Cells were lysed on ice in lysis buffer containing 10 mM Tris-HCl pH 7.8, 150 mM NaCl, 1 mM EDTA, 25 mM NaF, 1% (w/v) Nonidet P40, protease inhibitors (10 μg/ml aprotinin, 2.5 μg/ml leupeptin, and 1 mM phenylmethylsulfonyl fluoride), and 1 mM sodium orthovanadate (a phosphatase inhibitor). The lysates were cleared by centrifugation at 16,000g for 20 min at 4°C and protein concentrations in the lysates were measured (Bradford protein assay, Bio-Rad, Hercules, CA).
For immunoprecipitation, equal amounts of cell lysates were mixed with the indicated antibodies and incubated end-over-end at 4°C for 90 min. Protein G Sepharose beads (Amersham Biosciences) were added for 60 min and subsequently washed three times in ice-cold lysis buffer. Proteins were eluted and denatured in Laemmli sample buffer containing 5% β-mercapto-ethanol, separated by SDS–PAGE (15% polyacrylamide gels) and transferred to nitrocellulose membrane (Hybond ECL, Amersham Biosciences). Membranes were blocked in Tris-buffered saline with Tween 20 (TBS-T, 150 mM NaCl, 20 mM Tris-HCl, pH 7.5, 0.1% Tween 20) and with 5% non-fat dried milk overnight at 4°C. Immunoblotting was performed with the indicated primary antibodies diluted in TBS-T with 5% non-fat dried milk for 120 min at room temperature. After washing in TBS-T for 30 min, membranes were incubated with anti-rabbit IgG coupled to HRP in TBS-T with 5% non-fat dried milk for 60 min at room temperature. The antibody-antigen complexes were detected using the ECL system and visualized with a Lumi-Imager imaging analyzer (Roche, Basel, Switzerland). The intensity of bands was quantified by image analysis software (LumiAnalyst, Roche). In some cases, as indicated, blots were reprobed with an anti-actin antibody (Santa Cruz) for monitoring the quantity and integrity of protein.
Preparation of nuclear extracts and electrophoretic mobility shift assays (EMSA)
Nuclear extracts were prepared as previously described (Andrews and Faller, 1991) with minor modifications. Proteins were quantified and 15 μg was used for EMSA. Double-stranded oligonucleotide (Santa Cruz Biotechnology) containing the AP-1 consensus binding site (5′-CGCTTGATGACTCAGCCGGAA-3′) were end-labeled with [γ-32P]ATP (∼6,000 Ci/mmol, Amersham Biosciences) using T4 polynucleotide kinase (New England Biolabs). Nuclear extracts were incubated with labeled oligonucleotide in the absence or presence of 7.5 pmol (50-fold excess) unlabeled oligonucleotide in EMSA buffer (10 mM HEPES-KOH (pH 7.8), 50 mM KCl, 1 mM EDTA (pH 8.0), 5 mM MgCl2, 10% glycerol, 5 mM, dithiothreitol, 0.7 mM, 2 μg poly(dI-dC), and protease inhibitors). To identify components bound to the cognate sequence motif, antibodies against FosB or MafG were incubated with the gel shift reactions. DNA–protein complexes were resolved by electrophoresis on 4% polyacrylamide gels and visualized with a BAS-2000 Bio-Imaging Analyzer (Fuji film, Tokyo, Japan).
Immunofluorescence
CHO-K1 cells were transiently transfected with the cDNAs for FosB and MafG, and grown for 24 h before fixation. Cells were fixed in 1% paraformaldehyde for 20 min, paraformaldehyde groups were blocked in 10% normal goat serum in PBS for 10 min and the cells were permeabilized in 0.1% Tween-20 in PBS for 10 min. Thereafter, the cells were blocked in 5% FCS in PBS for 60 min and incubated with the primary antibodies of FosB and FLAG in 5% FCS in PBS for 120 min. After extensive washing in PBS, the cells were incubated with the appropriate fluorophore-labeled secondary antibodies. The preparations were mounted in 50% glycerol and images were taken with the VANOX-S inverted fluorescence microscope (Olympus, Tokyo, Japan).
Semiquantitative reverse transcription-PCR
Total cellular RNA from CHO-K1 cells was prepared with the commercial RNeasy kit (Qiagen, Hilden, Germany). Chromosomal DNA was removed by digestion with RNase-free DNase I (Qiagen). One microgram of total RNA was used for reverse transcription-PCR (RT-PCR) using OneStep RT-PCR system (Qiagen). PCR was performed under the following conditions for 30 cycles: denaturation at 94°C for 30 sec, annealing at 55°C for 30 sec, and extension at 72°C for 1 min. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used to normalize input RNA. Primers used for semiquantitative RT-PCR are as follows: MafG sense primer 5′- CTTGCAGAGTGCCTGCTCAC-3′ and antisense primer 5′-TCAAAGACCTGCCTGGCAA-3′ (655 bp); FosB sense primer 5′-ATGTTTCAAGCTTTTCCCGG-3′ and antisense primer 5′-TTACAGAGCAAGAAGGGAGG-3′ (1,017 bp); MMP-1 sense primer 5′-TTGTTGCTGCCCATGAGCTT-3′ and antisense primer 5′-ACTTTGTCGCCAATTCCAGG-3′ (639 bp) (Chen et al., 2003); c-Jun sense primer 5′-AAGAACTCGGACCTTCTCAC-3′ and antisense primer 5′-GGATCTGTTGGGGCAAGTGG-3′ (490 bp) (Xu et al., 2003); GAPDH sense primer 5′-ATGGTGAAGGTCGGCGTGAA-3′ and antisense primer 5′- TTACTCCTTGGAGGCCATGTA-3′ (1,008 bp) (Robbins et al., 1995). The PCR products were separated by electrophoresis on an agarose gel containing ethidium bromide and were visualized with a Lumi-Imager imaging analyzer (Roche). The intensity of bands was quantified by image analysis software (Roche).
Statistical analyses
Results are expressed as the intensity relative to the control value, with mean ± SEM. The significance of differences in intensity of antibody–antigen complexes was evaluated using the Mann–Whitney U-test. The probability level taken to indicate significance was P < 0.05.
RESULTS
Extracellular acidification induces expression of FosB and MafG
To investigate whether FosB and small Maf proteins (sMafs) are expressed in response to extracellular acidification in vitro, the extracellular pH of cultured CHO-K1, HEK293, and PC 12 cells was decreased from 7.40 to 6.60. After 60 min incubation, the expression of FosB and sMafs were assessed by immunoblotting analysis with anti-FosB antibody and anti-small Maf proteins (MafG/K/F) antibody (Fig. 1). Expression patterns of FosB and sMafs were increased by 337% and 235% (CHO-K1), 371% and 218% (HEK293), and 266% and 189% (PC 12) over control value (pH 7.40), respectively. From these observations, it appears that induction of FosB and sMafs by acidosis occurs in several mammalian cell types.

Acidosis-induced expression of FosB and small Maf proteins (sMafs) in cultured cells. A: Culture media of CHO-K1, HEK293, and PC 12 cells were replaced with fresh media adjusted to pH 7.40 or 6.60, and then the cells were incubated for additional 60 min. Each cell lysate (20 μg) was immunoblotted using anti-FosB (upper parts) and anti-MafF/G/K (middle parts). Representative blots from three replicates are shown. The positions of standard molecular masses (in kilodaltons, kDa) are indicated at the left. B: Histograms of the relative intensity of the H+-induced FosB (upper part) and sMafs (lower part). Intensity values of bands are expressed as a percentage of values from the intensity at extracellular pH 7.40. Results are averages of three individual experiments. Asterisks indicate a significant difference with the value at pH 7.40 (P < 0.05). pHo, extracellular pH.
On the basis of the above results, we planned to determine whether acidosis-induced expression of FosB and sMafs is regulated in a pH-dependent manner. The expression of FosB was increased with a decrease in extracellular pH in a pH-dependent manner (Fig. 2A, top part). Intensities of sMafs were also greater with a decrease in extracellular pH (Fig. 2A, middle part). Incubation in culture medium at pH 6.60 for 60 min produced a 3.9-fold and 2.2-fold increase in intensities of FosB and sMafs, respectively, compared with the values at pH 7.40 (Fig. 2B). The correlation coefficient (R) between extracellular pH values and the protein expression was −0.976 for FosB and −0.981 for sMafs. Taken together, these data show that the decrease in extracellular pH correlates well with expression of FosB and sMafs.

Extracellular acidification induces expression of FosB and sMafs with pH dependence. Culture medium of CHO-K1 cells was replaced with fresh medium adjusted to pH 7.40, 7.20, 7.0, 6.80, or 6.60, and cells were incubated for additional 60 min. A: Western blots were performed using the indicated specific antibodies. Representative blots from four replicates are shown. The positions of standard molecular masses (in kDa) are indicated at the left. B: Intensity values of bands are expressed as a percentage of the values of the intensity at pH 7.40. Results are averages of four individual experiments. Asterisks indicate a significant difference with the value at pH 7.40 (P < 0.05); (●), FosB; (▴), sMafs; pHo, extracellular pH.
We next examined kinetics of FosB and sMafs expression following a decrease in extracellular pH (Fig. 3). After replacement of the culture medium of CHO-K1 cells with fresh medium adjusted to pH 6.60, the cells were incubated for 0.5, 1, 2, 4, or longer than 8 h. Immunoblotting revealed that acidosis-induced expression of FosB and expression of sMafs were not greatly increased by 0.5 h of the stimulation but thereafter rose abruptly and reached a peak after 1–2 h of stimulation. After 8 h, the both FosB and sMafs levels were returned to slightly above control levels.

Time course of FosB and sMafs expression in response to extracellular acidification. The culture medium of CHO-K1 cells was replaced with fresh medium adjusted to pH 6.60, and cells were incubated for additional 0, 0.5, 1.0, 2.0, 4.0, or 8.0 h. A: Western blots were performed using the indicated specific antibodies. Representative blots from four replicates are shown. The positions of standard molecular masses (in kDa) are indicated at left. B: Intensity values of bands are expressed as a percentage of values from the intensity at 0 min. Results are averages of four individual experiments. Asterisks indicate a significant difference with the value at 0 min (P < 0.05); (●), FosB; (▴), sMafs; pHo, extracellular pH.
FosB and MafG co-localize in the nucleus and their heterodimer binds to an AP-1 sequence
To further address the role of the FosB and sMafs in acidosis, we investigated the nuclear co-localization between FosB and MafG by immunofluorescence. A previous report has demonstrated that gene expression of MafG is induced by acidosis following hypercapnic stimulation (Shimokawa et al., 2000). Therefore, we focused on MafG among other sMafs. Both FosB and MafG were expressed within the nucleus. When FosB was expressed together with FLAG-tagged MafG in CHO cells, the two proteins partially co-localized in the nucleus (Fig. 4A). Moreover, immunoprecipitation of FosB showed that MafG associates with FosB (Fig. 4B). These results indicate that FosB and MafG form heterodimers in the nucleus when they are co-expressed.

Evidence of heterodimerization of MafG with FosB. A: Nuclear co-localization between MafG and FosB. FosB and Flag-tagged MafG were transiently transfected into CHO-K1 cells. After 22 h, cells were subjected to immunofluorescence with rabbit polyclonal anti-FosB and mouse monoclonal anti-FLAG antibodies. FosB is visualized in green by Alexa Fluor 488-labeled secondary goat anti-rabbit and FLAG (MafG) is stained red with Alexa Fluor 568-conjugated goat anti-mouse antibodies. The merged pictures from the two channels are also shown. Scale bar: 10 μm. N, nucleus. B: Immunoprecipitants of MafG–FosB complex. FosB and Flag-tagged MafG were transiently transfected into CHO-K1 cells. After 48 h, cells were subjected to immunoprecipitation with the indicated antibodies.
To investigate whether FosB–MafG complex is able to bind to AP-1 sequence, both proteins were expressed in CHO-K1 cells and their DNA binding activities were examined by EMSA (Fig. 5). Single transfection of FosB did not bind to AP-1 probe (lane 1). Our result is consistent with the fact that the two Fos family proteins cannot form dimer (Kataoka et al., 1994). Co-expression of FosB and FLAG-tagged MafG fusion protein resulted in strong DNA binding (closed arrowhead, lane 2). Specificity of the DNA binding was confirmed by the addition of excess unlabeled AP-1 probe (lane 3). To confirm that the complex consists of FosB and MafG, we employed the anti-FosB and anti-FLAG antibodies in an EMSA. Incubation of the extracts from transfected cells expressed with each antibody resulted in a super-shifted complex (open arrowhead, lanes 4 and 5). Taken together, these results confirm that FosB and MafG complexes can bind to AP-1 site in the nucleus.

MafG forms heterodimers with FosB and bind to AP-1 site. FosB expression plasmid was transfected into CHO-K1 cells in the absence or the presence of Flag-tagged MafG expression plasmid. Resulting DNA binding activities were analyzed by EMSA with 32P-labeled AP-1 consensus binding site. To confirm binding specificities, excess unlabeled probe was included in the binding reaction mixtures. MafG-FosB heterodimers and AP-1 complexes are shown by closed arrowhead. Supershift complexes bound with anti-FosB or anti-FLAG antibody are indicated by open arrowhead.
Extracellular acidification increases DNA binding of FosB–MafG complex to AP-1 site
Because expression of FosB and sMafs was increased in response to extracellular acidification, we speculated that extracellular acidification enhances DNA binding activity of FosB-sMafs heterodimer to AP-1 site. To test this hypothesis, both proteins were expressed in CHO-K1 cells, and the extracellular pH of the cultured cells was decreased from 7.40 to 6.60. After 2 h incubation, dimerization and DNA binding activities between FosB and MafG were examined by immunoprecipitation and EMSA, respectively. FosB and MafG expressed by transfection were showed bound to each other at pH 7.40. The amounts of these complexes increased significantly when extracellular pH was shifted from 7.40 to 6.60 (Fig. 6A). The increase was observed about 260% at extracellular pH 6.60 compared with those at 7.40 (Fig. 6B). Similarly, acidic condition (extracellular pH 6.60) enhanced DNA binding activity of FosB–MafG complex to AP-1 sequence, compared with their counterparts at pH 7.40. Taken together our results clearly show that extracellular acidification significantly increases the DNA binding activity of FosB–MafG complexes to the AP-1 sequence and increases the expression of Fos and sMafs.

Effects of extracellular acidification to dimerization and DNA binding activity of MafG and FosB. CHO-K1 cells overexpressing FLAG-MafG and FosB, or FLAG were replaced with fresh medium adjusted to pH 7.40 or 6.60, and cells were incubated for additional 120 min. A: Western blots were performed using the indicated specific antibodies after immunoprecipitation. Representative blots from four replicates are shown. The positions of standard molecular masses (in kDa) are indicated at left. B: Histograms of the relative intensity of the acidosis-induced heterodimerization MafG with FosB. Intensity values of bands are expressed as a percentage of values from the intensity at extracellular pH 7.40 with transfected expression vector of FLAG-MafG and FosB. Results are averages of four individual experiments. Asterisks indicate a significant difference with the value at pH 7.40 (P < 0.05). C: DNA binding activity of MafG–FosB complex increases by extracellular acidification. CHO-K1 cells overexpressing FLAG-MafG and FosB were replaced with fresh medium adjusted to pH 7.40 or 6.60, and cells were incubated for additional 120 min. Resulting DNA binding activities were analyzed by EMSA with 32P-labeled AP-1 consensus binding probe. MafG-FosB heterodimers and AP-1 complexes are shown by closed arrowhead. pHo, extracellular pH.
Extracellular acidification induces MMP-1 expression
On the basis of the above results, we investigated whether extracellular acidification was associated with an activation of genes that having AP-1 sequence at the promoter region. Because it is known that transcriptional induction of matrix metalloproteinase-1 (MMP-1, collagenase-1) is regulated by AP-1 (Doyle et al., 1997), we assessed mRNA expression of MMP-1 during extracellular acidification. The extracellular pH of cultured CHO-K1cells was decreased from 7.40 to 6.60. After 1 or 12 h incubation, the mRNA expression of MafG, FosB, MMP-1, and c-Jun were assessed by RT-PCR (Fig. 7). The mRNAs expression level of MafG, FosB, and c-Jun were increased over control value (pH 7.40) after 1 h incubation and returned to slightly above to control levels after 12 h. These results correlate well with results of protein expression as shown in Figures 1 and 3. On the other hand, although extracellular acidification did not affect the MMP-1 expression after 1 h incubation, incubation in culture medium at pH 6.60 for 12 h produced a 2.5-fold increase in intensities of MMP-1, compared with the values at pH 7.40. These results suggest that extracellular acidification induces MMP-1 expression through stimulation of expression of immediately early genes (FosB and c-Jun) and MafG.

MMP-1 mRNA expression is induced by extracellular acidification. CHO-K1 cells were incubated in the indicated pH for 1 h (A) or 12 h (B). Total RNA was then isolated and used for RT-PCR as described in “Materials and Methods.” Intensity values of transcripts (MafG, FosB, MMP-1, and c-Jun) are normalized to GAPDH mRNA levels and expressed as a percentage of values from the intensity at extracellular pH 7.40 (lower parts of A and B). Results are averages of four individual experiments. Asterisks indicate a significant difference with the value at pH 7.40 (P < 0.05). pHo, extracellular pH.
DISCUSSION
Previous studies implicated gene expressions of mafG and mafG-2 in intracellular signal transduction during acidosis (Shimokawa et al., 2000;2001). In the present report, we describe that extracellular acidification leads to heterodimerization between MafG and FosB as well as activation of DNA binding activity of the MafG-FosB dimer. These effects were pH- and time-dependent (Figs. 2 and 3) and were observed in several cell lines (CHO-K1, HEK293, and PC 12, Fig. 1). Similarly, our previous study indicated that low pH medium (extracellular pH 6.60) promotes phosphorylation of c-Jun NH2-terminal kinase (JNK), and c-Jun expression in a pH- and time-dependent manner (Shimokawa et al., 2004), indicating that increasing expression of bZIP transcription factors (Fos and Jun) during acidosis seems to be a common scenario in most of cells.
Moreover, we have demonstrated that MafG and Fos form functional heterodimer that can bind to AP-1 sequence in the nucleus (Fig. 4). It is known that the AP-1 complexes recognize two cis-acting elements. One is the phorbol 12-O-tetradecanoate 13-acetate (TPA)-responsive element (TRE, TGAC/GTCA). The other is different from TRE, because a single nucleotide, guanylic acid, is inserted into a sequence TGACGTCA (cAMP-responsive element, CRE, Montminy et al., 1986). On the other hand, Maf protein recognizes two types of relatively long palindromic consensus sequences, TGCTGACTCAGCA and TGCTGACGTCAGCA (Kataoka et al., 1994). The middle parts of these Maf-binding sequences were completely identical with two AP-1 sequences (TRE and CRE), suggesting that MafG-FosB heterodimer can bind to the core sequence, TGACTCA. In fact, MafG-FosB heterodimer resulted in binding to TGACTCA sequence (Fig. 5), which may participate in the regulation of genes having AP-1 site.
As mentioned above (1) acidosis induces expressions of MafG and FosB, (2) MafG forms heterodimer with FosB and the complex recognizes the AP-1 sequence. Does acidosis influence to dimerization and DNA binding activity of MafG and FosB? To address this question, we examined dimerization and DNA binding activities between MafG and FosB after extracellular acidification. Interestingly, acidosis was found to cause not only gene expressions of MafG and FosB but also to promote dimerization MafG with FosB and activation of the DNA binding activity of MafG–FosB complex to AP-1 site (Fig. 6). To determine the further signaling pathway in response to extracellular acidification, we investigated mRNA expression of MMP-1, one of the genes that having AP-1 site. We detected the induction of MMP-1 mRNA following that of MafG and FosB (Fig. 7). This observation is not direct evidence for binding to AP-1 site in MMP-1 promoter of acid-induced MafG and FosB. However, from the results of our present study and a report that transcriptional induction of MMP-1 is regulated by AP-1 (Doyle et al., 1997), extracellular acidification seems to regulate genes that having AP-1 site through stimulation of FosB and MafG.
Regarding the physiological significance of induction to MafG/FosB and genes having AP-1 site by extracellular acidification, we have first considered apoptosis. In the case of hypoxia (1% O2, 5% CO2, balance N2 for 24 h), the expression of c-jun and c-fos mRNAs and up-regulation of DNA binding activity of AP-1 are also induced (Bae et al., 1998). The hypoxia-induced up-regulation is associated with apoptosis in human hepatocellular carcinoma cells (HepG2). Exposure of UV radiation and chemotherapeutic agents also induces an increase in DNA binding activity of AP-1 in connection with apoptosis (Goldstone and Lavin, 1994; Gillardon et al., 1994). A drastic change of extracellular environments (hypoxia) and genotoxic stress (UV and chemotherapeutic agents) would induce apoptosis. These observations suggest that up-regulation of gene expression, dimerization, and DNA binding activity of MafG and FosB during acidosis may be involved in inducing apoptosis. Moreover, MMPs secreted from myocardial cells can induce apoptosis with subsequent exacerbation of cardiac dysfunction (Heymans et al., 1999). Many tumor cells have relatively acidic extracellular pH and are killed by intracellular acid-induced injury. The acid-induced cell death depends on bax, a proapoptotic binding partner of bcl-2, and on JNK signaling pathways (Zanke et al., 1998). Recently, Yamamoto et al. (2000) reported that acidification of the cytoplasm using cycloprodigiosin hydrochloride (cPrG(HCl), a novel H+/Cl− symport drug, leads to apoptosis in cancer cells through up-regulation of Fas ligand, JNK, and caspases. In the present study, we have found no definitive relationship between extracellular acidosis and apoptosis in normal cells. DNA fragmentation is a reliable index of apoptosis. Though CHO-K1 cells were incubated in medium at pH 6.60 for 12 h, we could not detect the DNA ladder by agarose gel electrophoresis under these conditions (data not shown). Further studies will be needed to elucidate the induction of apoptosis by acidosis and its detailed signaling pathway.
Several reports have pointed the critical roles of the Maf family in the development and function in the brain. MafB is the avian homolog of the murine kreisler gene product and essential for hindbrain development (Cordes and Barsh, 1994). Recently, Blanchi et al. (2003) reported a critical function of MafB on respiratory rhythmogenesis. They have identified MafB as a marker of a specific subpopulation of neurons in the pre-Bötzinger complex, one of the principal sites of respiratory rhythmogenesis in the brainstem. Mice deficient for MafB (Mafb−/−) die from central apnea at birth, and in vitro preparations of Mafb−/− brainstems failed to generate a normal rhythmic discharge pattern. Together these observations and our data indicate that Maf proteins play critical roles in the adaptation to hypercapnia and respiratory rhythmogenesis. Several genes have been linked to human syndromes with various respiratory distresses, such as Prader–Willi syndrome (Ren et al., 2003), sudden infant death syndrome (SIDS, Kinney et al., 2003), and congenital central hypoventilation syndrome (Shirasawa et al., 2000). Maf proteins may thus provide a new insight into understanding of mechanisms for these diseases relating to respiration.
We have elucidated a part of intracellular signaling mechanism during acidosis, but it is still not clear what senses change in extracellular pH, and how it converts into an intracellular signal. Recently, some pH-sensing molecules respond to change of extracellular pH were identified. Waldmann et al. (1997) cloned the H+-gated cation channel (ASIC, for acid-sensing ionic channel) that belongs to the amiloride-sensitive Na+ channel/degenerin family of ion channels. ASIC is expressed in dorsal root ganglia and is also distributed widely throughout the brain. The H+-gated cation channel is activated transiently by rapid extracellular acidification and induces cation (Na+, Ca2+, K+) influx. More recently, it has been shown that ovarian cancer G-protein-coupled receptor 1 (OGR1), previously described as a receptor for sphingosylphosphorylcholine, acts as an H+-sensing receptor stimulating inositol phosphate (IP) formation (Ludwig et al., 2003). The receptor is stabilized in an inactive state at pH 7.8, and is fully activated at pH 6.8. Pertussis toxin did not inhibit IP formation measured at pH 7.0, indicating that OGR1 acts through Gq. Ovarian cancer G-protein-coupled receptor 4 (OGR4) also responds to pH changes, the receptor promotes cAMP formation through Gs. We succeeded in cloning of novel extracellular pH-dependent glucose transporter proton-associated sugar transporter-A (Past-A) from the brain of hypercapnia rats using differential display technique (Shimokawa et al., 2002). Past-A is able to facilitate glucose uptake during extracellular acidification. Increased expression of Past-A in hypercapnia may protect neurons from acidosis-induced cell damage by supplying extra energy source for recovery. Without such a protection mechanism the brain may be more fragile for metabolic acidosis followed by brain ischemia, trauma, and infection. With this regard, Past-A may play crucial role in maintaining homeostasis of brain. Further functional analysis of these pH-sensing molecules may provide new insights into the biochemical regulation of bZIP proteins during acidosis.
Acknowledgements
We thank Dr. Tohru Murakami for technical support of immunofluorescence. We are also thankful to other members of the department of integrative physiology for helpful discussions and critical comments on the study.

