

Extensive theoretical study on the low-lying electronic states of silicon monofluoride cation including spin-orbit coupling
Kun Liu
Beijing National Laboratory for Molecular Sciences, State Key Laboratory of Molecular Reaction Dynamics, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100080, People's Republic of China
Graduate University of the Chinese Academy of Sciences, Beijing 100049, People's Republic of China
Search for more papers by this authorCorresponding Author
Wensheng Bian
Beijing National Laboratory for Molecular Sciences, State Key Laboratory of Molecular Reaction Dynamics, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100080, People's Republic of China
Beijing National Laboratory for Molecular Sciences, State Key Laboratory of Molecular Reaction Dynamics, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100080, People's Republic of ChinaSearch for more papers by this authorKun Liu
Beijing National Laboratory for Molecular Sciences, State Key Laboratory of Molecular Reaction Dynamics, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100080, People's Republic of China
Graduate University of the Chinese Academy of Sciences, Beijing 100049, People's Republic of China
Search for more papers by this authorCorresponding Author
Wensheng Bian
Beijing National Laboratory for Molecular Sciences, State Key Laboratory of Molecular Reaction Dynamics, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100080, People's Republic of China
Beijing National Laboratory for Molecular Sciences, State Key Laboratory of Molecular Reaction Dynamics, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100080, People's Republic of ChinaSearch for more papers by this authorAbstract
Ab initio calculations on the low-lying electronic states of SiF+ are performed using the internally contracted multireference configuration interaction method with the Davidson correction and entirely uncontracted aug-cc-pV5Z basis set. The effects of spin-orbit coupling are accounted for by the state interaction approach with the full Breit–Pauli Hamiltonian. The entire 23 Ω states generated from the 12 valence Λ–S states, which correlate with the first dissociation channel are studied for the first time. Good agreement is found between the calculated results and the available experimental data. The spin-orbit coupling effects on the potential energy curves and spectroscopic properties are studied. Various curve crossings are revealed, which could lead to the predissociation of the a3Π, A1Π, and (2)3Σ+ states and the predissociation pathways are analyzed based upon the calculated spin-orbit matrix elements. The calculated ionization potentials of the ground-state SiF to a few states of SiF+ are in good agreement with the available experimental measurements. Moreover, the transition dipole moments of the dipole-allowed transitions and the transition properties for the A3Π0+–X1Σ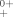 and B3Π1–X1Σ
and B3Π1–X1Σ transitions are predicted, including the Franck–Condon factors and the radiative lifetimes. © 2007 Wiley Periodicals, Inc. J Comput Chem, 2008
transitions are predicted, including the Franck–Condon factors and the radiative lifetimes. © 2007 Wiley Periodicals, Inc. J Comput Chem, 2008
References
- 1 Davis, D. N. Astrophys J 1947, 106, 28.
- 2 Davis, S. J.; Hadley, S. G. Phys Rev A 1976, 14, 1146.
- 3 Vasile, M. J.; Stevie, F. A. J Appl Phys 1982, 53, 3799.
- 4 Winters, H. F.; Houle, F. A. J Appl Phys 1982, 54, 1218.
- 5
Grill, A.
Gold Plasma in Materials Fabrication;
IEEE Press:
New York,
1994.
10.1109/9780470544273 Google Scholar
- 6 Lieberman, M. A.; Lichtenberg, A. J. Principles of Plasma Discharges and Materials Processing; Wiley: New York, 1994.
- 7 Winters, H. F. J Vac Sci Technol B 1983, 1, 927.
- 8 Haring, R. A.; Haring, A.; Saris, F. W.; deVries, A. E. Appl Phys Lett 1982, 41, 174.
- 9 Oostra, D. J.; Haring, A.; deVries, A. E. J Vac Sci Technol B 1986, 4, 1278.
- 10 Reents, W. D.,Jr.; Mujsce, A. M.; Bondybey, V. E.; Mandich, M. L. J Chem Phys 1987, 86, 5568.
- 11 Hayes, T. R.; Wetzel, R. C.; Baiocchi, F. A.; Freund, R. S. J Chem Phys 1988, 88, 823.
- 12 Petrmichl, R. H.; Peterson, K. A.; Woods, R. C. J Chem Phys 1988, 89, 5454.
- 13 Akiyama, Y.; Tanaka, K.; Tanaka, T. Chem Phys Lett 1989, 155, 15.
- 14 Weber, M. E.; Armentrout, P. B. J Chem Phys 1988, 88, 6898.
- 15 Fisher, E. R.; Kickel, B. L.; Armentrout, P. B. J Phys Chem 1993, 97, 10204.
- 16 Reid, C. J. Chem Phys 1996, 210, 501.
- 17 Bredohl, H.; Breton, J.; Dubois, I.; Esteva, J. M.; Macau-Hercot, D.; Remy, F. J Mol Spectrosc 1999, 195, 281.
- 18 Peterson, K. A.; Woods, R. C. J Chem Phys 1988, 89, 4929.
- 19 Peterson, K. A.; Woods, R. C. J Chem Phys 1990, 92, 6061.
- 20 Robbe, J. M. J Mol Spectrosc 1985, 112, 223.
- 21 Ricca, A.; Bauschlicher, C. W.,Jr. Chem Phys Lett 1998, 287, 239.
- 22 Karna, S. P.; Grein, F. Chem Phys Lett 1988, 150, 171.
- 23 Peterson, K. A.; Woods, R. C.; Rosmus, P.; Werner, H.-J. J Chem Phys 1990, 93, 1889.
- 24 Karna, S. P.; Grein, F. J Mol Spectrosc 1987, 122, 28.
- 25 MOLPRO, a package of ab initio programs designed by Werner, H.-J.; Knowles, P. J. Version 2002.6, with contributions from Amos, R. D.; Bernhardsson, A.; Berning, A.; Celani, P.; Cooper, D. L.; Deegan, M. J. O.; Dobbyn, A. J.; Eckert, F.; Hampel, C.; Hetzer, G.; Knowles, P. J.; Korona, T.; Lindh, R.; Lloyd, A. W.; McNicholas, S. J.; Manby, F. R.; Meyer, W.; Mura, M. E.; Nicklass, A.; Palmieri, P.; Pitzer, R.; Rauhut, G.; Schütz, M.; Schumann, U.; Stoll, H.; Stone, A. J.; Tarroni, R.; Thorsteinsson, T.; Werner, H.-J.
- 26 Werner, H.-J.; Knowles, P. J. J Chem Phys 1985, 82, 5053.
- 27 Knowles, P. J.; Werner, H.-J. Chem Phys Lett 1985, 115, 259.
- 28 Werner, H.-J.; Knowles, P. J. J Chem Phys 1988, 89, 5803.
- 29 Knowles, P. J.; Werner, H.-J. Chem Phys Lett 1988, 145, 514.
- 30 Laughoff, S. R.; Davidson, E. R. Int J Quantum Chem 1974, 8, 61.
- 31 Douglas, M.; Kroll, N. M. Ann Phys 1974, 82, 89.
- 32 Hess, B. A. Phys Rev A 1986, 33, 3742.
- 33 Woon, D. E.; Dunning, T. H.,Jr. J Chem Phys 1993, 98, 1358.
- 34 Dunning, T. H.,Jr. J Chem Phys 1989, 90, 1007.
- 35 Kendall, R. A.; Dunning, T. H.,Jr.; Harrison, R. J. J Chem Phys 1992, 96, 6796.
- 36 Berning, A.; Schweizer, M.; Werner, H.-J.; Knowles, P. J.; Palmieri, P. Mol Phys 2000, 98, 1823.
- 37 Le Roy, R. J. LEVEL7.7: A computer program for solving the radial Schrödinger equation for bound and quasibound levels; University of Waterloo: Chemical Physics Research Report No. CP-661, 2005.
- 38 Johns, J. W. C.; Barrow, R. F. Proc Phys Soc (London) 1958, 71, 476.
- 39 Dyke, J. M.; Lewis A. E.; Morris, A. J Chem Phys 1984, 80, 1382.
- 40 Dyke, J. M.; Hoopr, N.; Morris, A. J Electron Spectrosc Relat Phenom 2001, 119, 49.
- 41 Petsalakis, I. D.; Theodorakopoulos, G. Chem Phys 2000, 254, 181.
- 42 Xu, H.; Balasubramanian, K. Chem Phys Lett 1995, 237, 7.
- 43 Balasubramanian, K.; Xu, H. J Mol Spectrosc 1995, 171, 555.
- 44 Moore, C. E. Atomic Energy Levels; National Bureau of Standard: Washington, DC, 1971.
- 45 Okabe, H. Photochemistry of Small Molecules; Wiley-Interscience: New York, 1978.
- 46 Heaven, M. C. Chem Soc Rev 1986, 15, 405.

