

Exploring the binding mechanism of HDAC8 selective inhibitors: Lessons from the modification of Cap group
Abstract
The abnormal expression of histone deacetylase 8 (HDAC8) has been reported to associate with various cancer entities (colon, breast cancer, pancreas, etc.) as well as parasitic diseases, making HDAC8 gradually develop into an attractive and potential therapeutic target. Among the various design strategies of selective HDAC8 inhibitors (modification of Cap, Linker, or zinc binding group regions), the optimization of Cap region has aroused great interest among the researchers. However, the detailed information underlying how the modification of Cap region influences the inhibitory activities is still unclear, and in this study, compounds 2c, 3g, and 3n were selected to explore the differences in binding mechanisms brought by Cap modifications via various computational approaches at the atomic level. Five residues (Y293, H167, D254, D165, and M261) have a large difference in energy contributions to the constructed systems, and the subpocket formed by Y293 and M261 could interact with Cap groups, triggering the differences in the energy contributions of the residues (H167, D254, and D165) located in metal-catalytic center. In summary, the compounds 2c, 3g, and 3n were selected as molecular probes to explore the binding mechanism, and the residues (Y293 and M261) forming the subpocket should be paid special attention in the design and synthesis of novel selective HDAC8 inhibitors.
Abbreviations
-
- Cap
-
- capping group
-
- HATs
-
- histone acetyltransferases
-
- HDAC8
-
- histone deacetylase 8
-
- HDACi
-
- HDAC inhibitors
-
- MD
-
- molecular dynamics
-
- MM/GBSA
-
- Molecular Mechanics Generalized Born Surface Area
-
- sHDAC8Is
-
- selectivity of HDAC8 inhibitors
-
- ZBG
-
- zinc binding group
1 INTRODUCTION
Acetylation of lysine residues in histone, as a key molecular mechanism belonging to epigenetic modification, is a reversible biological process catalyzed by histone acetyltransferases (HATs) and histone deacetylases (HDACs), and the normal cells have the ability to balance the bioactivities of HATs and HDACs.1-3 With the continuous development of epigenetic mechanisms, more and more studies have indicated that abnormal expression of HDAC is involved in multiple pathological conditions (apoptosis, inflammation, cell differentiation, cell death, and neuroplasticity), which makes HDACs gradually become potential and attractive targets for the treatment of cancer, inflammation, and neurodegeneration.4-9 Based on the homology with the yeast HDACs sequence, human HDAC members are classified into four classes, three of which belong to the metalloproteins containing zinc ion (Classes I [HDAC1-3, and HDAC8], Classes IIa [HDAC4, 5, 7, and 9], Class IIb [HDAC6 and 10], Classes IV [HDAC11]).10, 11 Currently, the clinically approved HDAC inhibitors are broad-spectrum and inhibit number of HDAC isoforms without subtype selectivity. Despite the superior therapeutic advantages, the off-target effects and strong toxicity pose challenges and limitations for their clinical application,12-15 making selective HDAC inhibitors become a hotspot in drug discovery.
Among the subtypes in HDACs family, HDAC8 is unique in the protein crystal structure and the biological function, which has been structurally characterized.16, 17 HDAC8 is mainly expressed and distributed in the nucleus and cytoplasm, which can catalyze the histone and non-histone substrates, including cohesin, ERRα, and cortactin.18, 19 Despite belonging to the class I, various differences between HDAC8 and HDAC1-3 makes HDAC8 a unique member.20 Above all, HDAC8 is an X-linked protein without relying on any other co-complexes for its bioactivity, and the amino acids constructing HDAC8 active pocket contains a methionine, which is also different from other HDAC subtypes.16, 21 In addition, as the smallest subtype in the HDAC family, the size of the entrance of HDAC8 active pocket is larger than that of other subtypes.22 Currently, several studies have proven that the abnormal expression of HDAC8 is associated with various diseases, especially cancers, and the deactivation of HDAC8 via RNA interference will inhibit the proliferation of various human cancer cell lines.6, 16, 23, 24 Additionally, the aberrant expression of HDAC8 is related to the stage of tumor development, such as neural crest-derived neuroblastoma, and malignant childhood cancer neuroblastoma,25 and contribute the cell proliferation of liver tumor cell lines.26 Indeed, specific inactivation of HDAC8 will lead to the inhibition of cell proliferation, accelerate the cell differentiation, and reduce the clonogenic growth,19 such as lymphoma and leukemic cells.27, 28 Moreover, compared to pan-HDAC inhibitors, selective HDAC8 inhibitors exhibit good therapeutic effects in neuroblastoma models with high efficacy and low toxicity.18, 24, 29 Therefore, HDAC8 is a critical target for the design of isozyme-selective lead compounds.
Till now, considerable efforts have been made to design the selective HDAC8 inhibitors, and the synthesized inhibitors basically conform the pharmacophore model of traditional pan-HDAC inhibitors, namely cap region (Cap), linker moiety (Linker), and zinc-binding group (ZBG) (Figure S1).16, 17, 30-32 There are various modification strategies for the selective HDAC8 inhibitors, and increasing studies have pointed out that the selective inhibitory activity of the small molecules against HDAC8 is sensitive to the Cap modification.24, 25, 33, 34 Thus, it is necessary to explore how the Cap modifications affect the binding mechanism of the inhibitors to HDAC8 at the atomic level.35 Based on the study,36 compounds 3g and 3n significantly enhanced their inhibitory activity against HDAC8 by modifying the Cap region compared to compound 2c (Figure S2), which could be used as molecular probes to explore the difference in the binding mechanism brought by Cap modification.37
In this study, molecular docking and molecular dynamic (MD) simulation were adopted to study the difference in inhibitory mechanism of the selected compounds in HDAC8.39-43 First, the complexes of HDAC8 with compounds 2c, 3g, and 3n were constructed, and their binding models were compared. Then, the simulation results were evaluated by the experimental values, and the key residues contributing to the ligands’ binding were identified.44, 45 Finally, the influences brought by the modification of Cap group on target-ligand interactions were analyzed and key residues with higher energy contribution to the selected compounds’ binding were discovered. In sum, the mechanism underlying how the Cap modification enhanced the inhibitory activity was discovered by decomposition free energy contribution among the key amino acids consisting of the binding pockets, which could provide valuable hints for the discovery of new HDAC8 inhibitors.
2 METHODS AND MATERIALS
2.1 Preparation of the systems
Before the simulation, the initial complex conformations ought to be obtained by the molecular docking approach via Glide embedded in Maestro mode.46 The molecular structures of the selected compounds were prepared by ChemBioDraw Ultra 14.0,47 and then were preprocessed by the LigPrep.48 Then, the protein receptor should also be prepared (adding hydrogen atoms, assigning the partial charges and protonation states, and minimizing the whole structure) before molecular docking using Protein Preparation Wizard.49 At present, the three-dimensional structure of human HDAC8 with inhibitor—Trichostatin A (TSA) (PDB entry: 1T64)38 can provide valuable instructional suggestion when determining the active binding pocket. Thus, on the basis of the spatial position of TSA in the PDB structure (1T64), the docking grid could be defined by the Receptor Grid Generation tool. Then, Glide docking was applied to determine the initial structures of the studied systems, and there were 5000 poses formed during the process of molecular docking, of which the best 400 conformations were selected after the energy minimization. The obtained docking poses, with the most similar orientation with TSA in spatial, were chosen as the initial binding poses.
2.2 Molecular dynamics simulation
Molecular dynamics (MD) simulations of all the prepared systems were conducted by GPU-accelerated PMEMD in AMBER1450 and the ligand and the protein were processed by AMBER ff14SB51 and general Amber force fields,52 respectively. Based on the previous studies,53, 54 the Li/Merz ion parameters with SPC/E water model were selected in this study,53-55 and the charges of the selected compounds 2c, 3g, and 3n were processed by antechamber.56 Besides, Gaussian 09 was used to optimize the geometry and calculate the electrostatic potential at HF/6-31G* level.57 Before the MD simulation, the three prepared systems should first experience energy minimization via the following two procedures: (1) applying harmonic restraint on solute atom (force constant= 10 kcal·mol−1·Å−2); (2) releasing atoms to move freely. During the process of energy minimization, the steepest descent approach and conjugated gradient method were applied, and then, the complexes were heated from 0 K gradually to 300 K by two steps (0 to 100 K and 100 to 310 K). During the heating of the systems, the protein should be restrained in the NVT ensemble, and 5 ns unrestrained equilibration at 310 K was conducted to equilibrate the periodic boundary conditions of the prepared systems. Finally, 500 ns unrestrained MD simulation was performed for the prepared complexes in the NPT ensemble (310 K and 1 atm). Langevin dynamics and Monte Carlo barostat58 were used to control the constant temperature and pressure, and the long-range electrostatic interactions were processed by Particle-mesh Ewald.59 All the analyses of MD trajectories (RMSD, representative conformation, and decomposition free energy) were conducted by cpptraj and mm_pbsa.pl programs embedded in AMBER14,58 and the 3D visualization was performed by PyMOL software.60
2.3 Calculation of the binding free energy
 (1)
(1) (2)
(2)The definition of 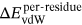 ,
, 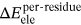 , and
, and 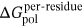 was similar to that in Eq. 1, and
was similar to that in Eq. 1, and 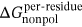 was calculated by recursively approximating a sphere around given atom based on the icosahedron (ICOSA) method.
was calculated by recursively approximating a sphere around given atom based on the icosahedron (ICOSA) method.
2.4 Identification of the key residues contributing the compounds’ binding through hierarchical clustering analysis
- (1)
Energy contributions of certain residues (≠0.1 kcal/mol) in all the prepared systems would be used to generate the 4-dimensional vector, and the similarities among the vectors were calculated using Manhattan distance with dist function embedded in R environment.
where i represented every dimension of the each residue energies a and b. (3)
(3) - (2)
Ward's minimum variance approach was adopted to minimize the total variance within the cluster using hclust function.
- (3)
The online tree generator iTOL58 was used to generate the hierarchical tree graph. The residues with higher decomposition free energy that favored the ligand-receptor interactions were shown in red color, and the residues displayed in blue hampered HDAC8 binding.
3 RESULTS AND DISCUSSION
3.1 Construction of the three studied complexes
Based on human HDAC8 crystal structure complexed with TSA (1T64)38 available in Protein Data Bank (PDB),62 molecular docking approach was adopted to construct the initial conformations of the compounds 2c, 3g, and 3n in HDAC8, and the similarity to the spatial orientation of TSA in the original crystal and the docking score could provide insights for the selection of the initial conformations. As illustrated in Figure 1, the active binding pocket of the three prepared systems primarily consisted of several flexible loops, and the conformation of the docked small molecules is consistent overall in space, especially Linker and ZBG region in the active pocket. In addition, the difference in all the superimposed studied complexes is mainly reflected in the interactions between the Cap groups and the different loop regions, which may be the main reason that induced the difference in their inhibitory activities. Certainly, the various interactions between the Cap region and the corresponding loop domains required further research and demonstration by molecular dynamic simulation.

3.2 Assessing the stability of the molecular dynamic simulation by RMSD analysis
The prepared complexes were further assessed by 100 ns MD simulation, and RMSD values of the skeleton atoms of the receptor protein, the residues consisting of the binding site, and the three selected compounds (2c, 3g, and 3n) were selected to monitor the dynamic equilibrium. According to the RMSD values (Figure S3), the prepared systems were able to reach equilibrium with slight fluctuation at 30 ns. Furthermore, to better verify the overall stability of the three systems, 70 ns MD production was conducted and showed that all the systems basically kept continuous and stable equilibration.
3.3 Verifying the MD results based on experimental data
MM/GBSA method was utilized to calculate the BFEs of all the prepared complexes, and the values of the BFEs were −27.35, −38.49, and −40.21 kcal/mol, respectively. In addition, based on the formula ∆Gexp = RT ln(IC50), the estimated binding affinities could be deduced from the IC50 values, and the values calculated by the formula were −8.30, −10.05 and −10.29 (as shown in Table 1).36 In this study, the system of HDAC8 complexed with compound 3n was selected as the reference to calculate the relative BFEs of all the prepared systems (∆∆GMM/GBSA) and the experimental results (∆∆Gexp). All the calculated values could be available in Table 1, and the two columns of data had the same ascending trend. The high correlation between the relative BFEs (∆∆GMM/GBSA) and the experimental results (∆∆Gexp) indicated the good reproducibility and reliability of the MD simulation results,63-66 which could be adopted to verify the results of the prepared complexes. In addition, ∆EvdW and ∆Eele were found to favor the compounds’ binding, while ∆Epol was discovered to hinder the selected compounds’ binding to HDAC8.
| Compounds | Calculated values | Experimental values | |||||||
|---|---|---|---|---|---|---|---|---|---|
| ∆Eele | ∆EvdW | ∆Gpol | ∆Gnonpol | ∆GMM/GBSA35?>a | ∆ ∆Gcalc35?>b | ∆Gexp35?>c | ∆ ∆Gexp35?>b | IC5035?>d | |
| 2c | −20.98 | −30.26 | 28.11 | −4.21 | −27.35 | 12.86 | −8.30 | 1.99 | 1400 |
| 3g | −31.06 | −36.69 | 34.27 | −5.02 | −38.49 | 1.72 | −10.05 | 0.24 | 82 |
| 3n | −30.78 | −32.30 | 27.69 | −4.83 | −40.21 | 0 | −10.29 | 0 | 55 |
- a Calculated MM/GBSA binding free energies in this study.
- b ΔΔG is defined as the change of binding free energy using compound 3n as a reference.
- c Estimated binding affinities based on IC50 values by the equation ∆Gexp = RT ln(IC50), where R = 8.314 J/(mol·K) and T = 310.0 K.
- d IC50 values obtained from the previous study.24
3.4 Identification of the key residues for target-ligand interactions
To quantify the key amino acids contributing to the interaction between the selected compounds and HDAC8, the amino acids with higher decomposition free energy (≥0.1 kcal/mol) to the compounds’ binding were discovered.67 As illustrated in Figure 2, the energy contribution of the same amino acid in HDAC8 to different compounds’ binding varied greatly (taking ASP254 in HDAC8 as an example, it contributed −0.2, −2.3, −1.5 kcal/mol to the binding of compound 2c, compound 3n, compound 3g, respectively), and different amino acids had different decomposition free energy contribution (taking compound 2c in HDAC8 as an example, the energy contribution of GLY138 equaled to − 2.04 kcal/mol, nearly 13 times of LEU21's contribution (−0.16 kcal/mol)). Per-residue energy contributions of the residues in HDAC8 to the binding of compound 2c, 3n, and 3g were hierarchically clustered, and the key amino acids were further identified. The residues with energy contribution were divided into six chains (Figure 3), and chain A was further divided into A1 and A2. As shown in Figure 6, the amino acids clustered into chain A2 had a large difference in the energy contribution for the selected compounds’ binding, which may be the main reason for the reduced inhibitory activity of compound 2c.


TYR293 belonging to chain A1 and all the amino residues clustered to chain A2 (HIE167, ASP254, ASP165, and MET261) had a relatively large difference in energy contribution to the binding of compounds 2c, 3g, and 3n (≥−04 kcal/mol). Furthermore, HIE167, ASP254, and ASP165 were located in the catalytic center surrounding the zinc ion, which played a vital role in maintaining the stability of zinc ion in the catalytic center (Figure 4A). HIE167, ASP254, and ASP165 had stronger interactions with compounds 3g and 3n compared to compound 2c, and due to the stronger interaction, the spatial position of the abovementioned amino acids would be affected, which would disturb the stability of the catalytic center (Figure 4B). Loop α and loop β formed a subpocket, and TYR293 in loop α and MET261 belonging to loop β had larger energy contributions for compound 3g and compound 3n's binding. The interactions between the Cap and the subpocket could maintain the stable active conformation, which was related to the Cap orientation (Figure 5). Certainly, in addition to chain A2, there were some energy differences between the other groups, but the difference was small and the complementary energy formed between the groups would offset these differences.


To further investigate the molecular mechanism of the decline in the inhibitory activity of compound 2c (more than 17-fold), an additional 400 ns molecular simulation was conducted on the HDAC8-2c system for a total of 500 ns. Based on Figure 6, there were relatively large fluctuations at the binding site, which indicated that the binding conformation might have changed. To further monitor the interaction between the zinc ion and ZBG region, the distance between the zinc ion and the carbonyl oxygen of ZBG of compound 2c was monitored, and the average distance was about 5.58 Å, which exceeded the reasonable interacting distance. However, the average distance between the Zn and the carbonyl oxygen of ZBG of compounds 3n and 3g in HDAC8 was 2.19 and 2.17 Å, which guaranteed their competition with zinc ions in the catalytic center (Figure 7). Moreover, the representative snapshot of this system was extracted every 50 ns to monitor the conformational changes. ZBG region of the traditional HDAC inhibitors should reach the bottom of the active pocket, which is the general common active conformation via chelating the zinc ion in the catalytic center, namely ZBG-IN. Based on the conformation of the extracted snapshots, the ZBG region of compound 2c has a tendency to gradually move away from the catalytic center of zinc ion as the function of computational time, and finally formed an inactive conformation (ZBG-OUT) (Figure 8). Although the compound 2c was relatively stable over 100 ns and maintained the active conformation (ZBG-IN), there was no guarantee that the small molecule would always stay the active conformation in the binding pocket. During the subsequent 400 ns MD simulation, the spatial conformation of compound 2c was reversed, especially the ZBG region, which was far away from the catalytic center. Therefore, in this studied system, the large-scale molecular dynamic simulations could better reflect the interactions between the compounds in the HDAC8.



4 CONCLUSION
Various computational methods were integrated in this study to research the binding mechanisms of the selected compounds (2c, 3g, and 3n) with different inhibitory activities at atomic level, which was applied to illustrate how Cap modification affected the selective inhibitory activity of compounds against HDAC8. First, four residues (GLY138, PHE139, PHE195, and TYR293) in HDAC8 were identified as the key residues with much higher energy contribution favoring the ligands’ binding. Due to the difference in the Cap groups, compounds 3g and 3n with rigid Caps could interact with TYR293 and MET261 located in the subpocket formed by loop α and loop β, and the traction effects of the subpocket on Cap maintained the stability of active conformation (ZBG-IN). The active conformation made the ZBG penetrate into the active pocket and interact with the residues around the zinc ion, which disturbed the stability of the catalytic center. However, compound 2c with flexible Cap group couldn't maintain active conformation (ZBG-IN) in HDAC8 during the long-time MD simulation due to the lack of the interactions between the Cap and the subpocket. Therefore, the HDAC inhibitors with large and rigid Cap groups could enhance their binding affinities with the loops consisting of the binding site and maintain the active conformations, which could provide valuable hints for the discovery of new drug candidates based on selective HDAC8 inhibitors.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
DH and YZ conceived the work and directed the experiment; YZ performed the whole simulation; YZ and JBY collected and confirmed protein and ligand structural data; YZ performed the subsequent analysis. DH and YZ drafted the manuscript. All authors read, edited and approved the final version of the manuscript.
Open Research
DATA AVAILABILITY STATEMENT
All data generated or analyzed during this study are included in this published article.

