

Nitric oxide-mediated inhibition of NFκB regulates hyperthermia-induced apoptosis
Abstract
Ascertaining the upstream regulatory mechanisms of hyperthermia-induced apoptosis is important to understand the role of hyperthermia in combined modality cancer therapy. Accordingly, we investigated whether (i) hyperthermia-induced apoptosis is mediated through the nitric oxide (NO) signaling pathway and (ii) inhibition of post-translational modification of IκBα and down regulation of NFκB-DNA binding activity is an intermediate step in NO-dependent apoptosis in MCF-7 breast cancer cells. For hyperthermia treatment, the cells were exposed to 43°C. Intracellular NO levels measured by the fluorescent intensity of DAF-2A and iNOS expression by immunobloting revealed an increased level of iNOS dependent NO production after 43°C. Apoptosis measured by Annexin V expression and cell survival by clonogenic assay showed a 20% increase in apoptosis after 43°C treatments. EMSA analysis showed a dose-dependent inhibition of NFκB-DNA binding activity. The hyperthermia-mediated inhibition of NFκB was persistent even after 48 h. Inhibition of NO by L-NAME rescued the NFκB-DNA binding activity and inhibits heat-induced apoptosis. Similarly, over-expression of NFκB by transient transfection inhibits heat-induced apoptosis. These results demonstrate that apoptosis upon hyperthermia exposure of MCF-7 cells is regulated by NO-mediated suppression of NFκB. J. Cell. Biochem. 106: 999–1009, 2009. © 2009 Wiley-Liss, Inc.
Hyperthermia treatment (HT) is an experimental cancer treatment modality. The scientific basis for HT is sound and the mechanisms of action for HT seem to be multifactorial. The usefulness of HT alone in treating advanced cancers [Herman et al., 1988, 1989; Jones et al., 2002] including prostate cancer [Szmigielski et al., 1988], tumors of the liver [Petrovich et al., 1988], brain tumors [Winter et al., 1985], ocular tumors [Coleman et al., 1988] and head and neck tumors [Shimm et al., 1988] has been indicated. However, HT in combination with ionizing radiation (IR) therapy achieves superior local control for several tumors [Lindholm et al., 1987]. The effect of HT combined with IR is due partly to its independent cytotoxic effects, and partly to its radiosensitizing effect [Overgaard, 1989]. Numerous clinical trials have clearly demonstrated that local HT added to RT, benefit patients (breast, neck nodes and malignant melanoma) with both improved local control [Lindholm et al., 1987; Valdagni et al., 1988; Overgaard et al., 1995; Vernon et al., 1996] and survival [van der Zee et al., 2000]. However, attainment of uniform temperature within the target tumor tissues is difficult to achieve [Sherar et al., 1997] and has been demonstrated that the size of tumor (local control rate is 70% in <16 cm2 tumors compared to 45% in >16 cm2 tumors) influenced the result. Because of the varied responses from patient to patient, type of tumor, tumor site, and tumor volume [Vernon et al., 1996], a firm conclusion could not be drawn regarding the long-term clinical effectiveness of HT [Perez et al., 1989, 1991; Kapp et al., 1990; Engin et al., 1993]. As the evidence for the value of adjuvant HT combined with IR in breast tumor control continues to accumulate, the detailed mechanisms of heat-induced cytotoxicity or apoptosis remain to be clearly elucidated [Ma et al., 2004]. Delineating the molecular mechanism(s), which can occur in cancer cells in response to HT, might shed light into the understanding of these varied responses due to HT.
Activation of NFκB has been implicated in stabilizing tumor cell growth by protecting the cells from undergoing apoptosis. Cell lines lacking the NFκB subunit p65, and those expressing a dominant negative IκBα, markedly augment TNFα induced apoptosis [Ghosh et al., 1998]. It has been recently reported that HT-induced apoptosis is regulated by heat-induced proteasome inhibition mediated down-regulation of NFκB-DNA binding activity [Pajonk et al., 2005]. NFκB is a heterodimer or homodimer of Rel family subunits p50, p52, p65/RelA, c-Rel, and Rel-B. A critical aspect of the regulation of NFκB activity is its posttranslational cytoplasmic sequestration by its major inhibitors, IκBs (IκBα, IκBβ, IκBγ, Bcl-3, p100, and p105). It is sequestered in the cytosol by inhibitor molecules of IκB family (IκBα, IκBβ, IκBγ, Bcl-3, p100, and p105). Activation of this pathway is normally achieved by phosphorylation of one of the most important inhibitors, IκBα, at two serine sites (ser 32 and ser 36) by IκB kinases. Proteasomal degradation of IκBα frees NFκB for translocation to the nucleus and activation of its target genetic programs [Baeuerle and Baltimore, 1996].
In addition, it has also been demonstrated that HT-induced apoptosis was also associated with intracellular generation of superoxide (O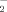 ) [Cui et al., 2006]. Although the involvement of excess intracellular oxidative stress is inferred in HT-induced apoptosis, the roles of the modulation of intracellular oxidative stress need further investigation. Nitric oxide (NO), a labile gaseous free radical, plays an important role in a wide range of physiological and pathological conditions. Large amounts of NO produced by inducible nitrous oxide synthase (iNOS) are harmful to tissues by producing peroxynitrite, which is a reaction product between NO and superoxide. iNOS gene expression is strictly controlled primarily at the transcriptional level [Cattell, 2002]. The promoter region of human iNOS contains at least one NFκB consensus site [Beck and Sterzel, 1996]. Taking into consideration HT-induced oxidative stress, we hypothesized that HT-induced apoptosis in human adenocarcinoma (MCF-7) breast cancer cells is mediated through the NO signaling pathway. Furthermore, the effects of this intracellular NO production on the inhibition of post-translational modification of IκBα and down regulation of NFκB-DNA binding activity, a possible mechanism involved in the process of HT-induced apoptosis were studied.
) [Cui et al., 2006]. Although the involvement of excess intracellular oxidative stress is inferred in HT-induced apoptosis, the roles of the modulation of intracellular oxidative stress need further investigation. Nitric oxide (NO), a labile gaseous free radical, plays an important role in a wide range of physiological and pathological conditions. Large amounts of NO produced by inducible nitrous oxide synthase (iNOS) are harmful to tissues by producing peroxynitrite, which is a reaction product between NO and superoxide. iNOS gene expression is strictly controlled primarily at the transcriptional level [Cattell, 2002]. The promoter region of human iNOS contains at least one NFκB consensus site [Beck and Sterzel, 1996]. Taking into consideration HT-induced oxidative stress, we hypothesized that HT-induced apoptosis in human adenocarcinoma (MCF-7) breast cancer cells is mediated through the NO signaling pathway. Furthermore, the effects of this intracellular NO production on the inhibition of post-translational modification of IκBα and down regulation of NFκB-DNA binding activity, a possible mechanism involved in the process of HT-induced apoptosis were studied.
MATERIALS AND METHODS
Cell Culture
Estrogen receptor positive human adenocarcinoma (MCF-7) breast cancer cell line was obtained from Dr. Beverly Teicher (Dana Farber Cancer Institute, Boston, MA). The cells were maintained as monolayer cultures by weekly serial passage in 100 mm tissue culture plates in Dulbecco's Modified Eagle medium (Life Tech., Grand Island, NY) with 44 mM sodium bi-carbonate, 4 mM L-glutamine, supplemented with 10.2 IU/ml penicillin/10.2 µg/ml streptomycin, and 10% heat-inactivated fetal bovine serum (JRH Tech., Lenexa, KS). When in exponential growth, the cells were observed to have a doubling time of approximately 16–18 h. For passage and for all experiments, the cells were detached using trypsin (0.25%)/EDTA (1%), re-suspended in complete medium, counted electronically using a Coulter Counter (Model ZBI, Coulter Electronics, Hialeah, FL) and incubated in a 95% air/5% CO2 humidified incubator. For all the experiments the cells were serum-starved by incubating at 2% serum for at least 16 h unless otherwise specified.
Hyperthermia Treatment
Cells in T25 or T75 flasks with plug-seal cap were tightly sealed by wrapping the cap with parafilm. The flasks placed in metal racks were totally immersed in a water bath at 37°C for 5 min. They were then immediately transferred into and totally immersed in a 43°C water bath for 15, 30, 45, or 60 min. At the end of incubation, the flasks were immediately transferred back into the 37°C water bath for 5 min. The temperature of the baths remained constant during this time. Each flask was then removed from the rack and wiped thoroughly. After removing the parafilm the flasks were placed in the 37°C incubator for continued incubation. For 37°C water bath controls, the flasks containing control cells were immersed in water bath maintained at 37°C for the whole duration when the hyperthermia treatment flasks were immersed at the adjacent 42°C water bath.
Electrophoretic Mobility Shift Assay
Nuclear protein extraction and electrophoretic mobility shift assay were performed as described in our earlier studies [Natarajan et al., 2002; Aravindan et al., 2008a,b]. For the competition assay, the nuclear extract was pre-incubated with unlabeled homologous NFκB oligonucleotide followed by addition of [γ-32P]-ATP labeled NFκB probe.
Immunobloting
Total protein extraction and western blot analysis was performed as described in our earlier studies [Natarajan et al., 2002]. For this study, the protein transferred membranes were incubated with either mouse anti-IκBα antibody (Santa Cruz Biotech, Santa Cruz, CA) or rabbit anti-iNOS antibody (Affinity Bioreagents Inc., Golden, CO). Blots were stripped and reblotted with anti-α-tubulin antibody (gift from Dr. Ashok Banergie, Department of Biochemistry, UTHSCSA, TX) to determine equal loading of samples. Stripping was accomplished using Re-Blot Western Blot Recycling Kit (Chemicon International, Inc., Temecula, CA).
NO Estimation
Intracellular NO level was determined using fluorescent nitric oxide indicator DAF-2DA [Hamuro et al., 2002]. Cells incubated for 1 h with a NO donor (SNP, 1 mM), was used as a positive control. The cells were harvested by trypsinization, washed twice with HBSS and the fluorescence intensity was measured by Fluorescein Activated Cell Sorter (FACS) at excitation wavelength of 488 nm and emission wavelength of 530 nm. The Cell Quest software (Becton, CA) was used for analysis.
Apoptotic Assay
Annexin V expression was quantitated using Annexin V-FITC Apoptosis Detection Kit I (Pharmingen, San Diego, CA). After treatment the cells were washed twice with ice cold PBS and re-suspended in binding buffer at a concentration of 1 × 106 cells/ml. 100 µl of cell suspension was added with 5 µl of Annexin V and 2 µl of propidium iodide, mixed gently and incubated at 22°C for 15 min in dark. Cells were re-suspended in 400 µl of binding buffer and analyzed immediately by FACS.
Transient Transfection
Transient transfection was carried out by the lipofection method using Effectene™ reagent (Qiagen, Inc., Valencia, CA). MCF-7 cells (1 × 106 cells/ml) were plated in 100 mm culture dishes and incubated at 37°C in 5% CO2/air incubator. When 80% confluent, the cells were serum starved by incubating at least for 16 h in complete medium containing 2% serum. Two micrograms of plasmid DNA (pCNA3 containing full-length expression of NFκB p65 and p50) were mixed with 100 µl of transfection reagent and incubated for 10 min at room temperature. The DNA-lipid complex was then slowly added to the cells and incubated for 2 h. The transfection medium was replaced with 10 ml of complete growth medium containing 2% serum. After 24 h recovery period, the cells were exposed to HT or treated with positive control. Over expression of NFκB p65 and p50 was monitored by immunoblotting using goat anti-human p65 and p50 primary antibodies (Santa Cruz Biotech.) and HRP-conjugated secondary antibody (Amersham).
Clonogenic Assay
For colony forming assay, cells (at a density of 300 cells/flask) seeded in a T25 tissue culture flasks were exposed to 43°C for 15, 30, or 60 min and incubated for 10 days. At the end of incubation period, colonies were fixed and stained with crystal violet and methylene blue dissolved in 90% ethanol and, counted manually under low magnification. The number of colonies was divided by the number of cells plated to calculate the colony formation efficiency.
Statistical Analysis
Data were expressed as mean ± SEM unless mentioned otherwise in the text. Statistical significance was assessed by one-way analysis of Variance (ANOVA) with Tukey's post hoc correction. A P value of <0.05 was considered as significant.
RESULTS
Dose-Dependent Inhibition of NFκB-DNA Binding Activity
To examine the effect of HT on the NFκB-DNA binding activity, MCF-7 cells were exposed to 43°C for 15, 30, 45, or 60 min. After HT, cells were incubated in a CO2/air incubator for additional 4 h. An autoradiogram showing the gel pattern is presented in Figure 1A. Inhibition of NFκB appears to be present after the cells are exposed to 43°C for 15 min compared to the control (cells exposed to 37°C). This HT-induced inhibition is clearly increased with a prolongation of heating period (43°C for 30, 45, and 60 min) and reached an utmost inhibitory level after 60 min, the maximum time point examined in this study. Densitometric analysis revealed a significant (P < 0.001) inhibition of NFκB-DNA binding activity compared to control level, after HT for 15, 30, 45, and 60 min, respectively (Fig. 1B). To confirm that the shifted band seen in EMSA analysis was due to the specific binding of NFκB to its sequence-specific oligonuleotide, a competition binding assay was performed. Nuclear extracts obtained from cells exposed to 43°C for 15 min and harvested at 2 h post-exposure were pre-incubated in the presence or absence of homologous unlabeled (cold) oligonucleotide competitor identical to the NFκB specific probe. In the presence of excess cold competitor, the γ-32P-labeled oligonucleotide will present only a few binding sites to NFκB, thereby reducing the intensity of the resulting band on the gel. As shown in Figure 1C, the NFκB-DNA binding activity was competitively reduced to 47% and 36.4% by the addition of 0.02 and 0.2 pmol of homologous unlabeled NFκB specific-double stranded oligonucleotide, respectively. The competitive inhibition of the DNA binding activity confirmed NFκB-specific binding.

A: A representative autoradiogram demonstrates the inhibition of NFκB-DNA binding activity upon exposure to an increasing duration at 43°C. Cells were incubated at 37°C or exposed to 43°C for 15, 30, 45, or 60 min. After HT, cells were incubated in a CO2/air incubator for additional 4 h. Nuclear extracts were analyzed by EMSA using γ-32p[ATP] labeled NFκB-specific probe. To visualize the reduced signals (i.e., less than the levels seen at 37°C), autoradiograms were overexposed. B: Densitometric analysis of NFκB-DNA binding activity. Quantitation of the levels of NFκB activation was performed by phosphor imaging using BioRad Phosphor Imaging System (GS-525, BioRad, Hercules, CA) and a BioRad Multi-Analyst software package with an integrated density program. C: Specificity of DNA-binding activity of HT-induced NFκB. Nuclear protein obtained from MCF-7 cells exposed to 43°C for 15 min and harvested after 2 h was incubated in the absence (lane 1) or presence (lanes 2 and 3) of increased concentrations of homologous unlabeled competitor for 5 min, and then probed with [γ-32P]-ATP labeled NFκB-specific oligonucleotide. The arrowhead indicates the specific DNA-binding activity of NFκB comprised of the p50/p65 heterodimeric complex. The fast moving lower band consists of the p50/p50 homodimer NFκB subunit complex. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Sustained Inhibition of NFκB-DNA Binding Activity
To determine whether the inhibition of NFκB-DNA binding activity was persistent or rapidly reversible process, the DNA-binding activity was examined at 10, 30 min, 1, 3, 6, 12, 24 and 48 h after HT for one hour (Fig. 2A). The data averaged for three independently treated flasks for each time point are presented in Figure 2B. The level of NFκB-DNA binding activity inhibition at each time point was compared to the control (37°C) for that incubation time. At all time points examined, the control showed little variation in NFκB-DNA binding activity. Compared to the control, HT for one hour profoundly suppressed the NFκB-DNA binding activity at 10 and 30 min after treatment. Similarly, we observed a relatively marked suppression in HT treated cells after 1, 3, 6, 12, and 24 h. Furthermore, HT treatment completely suppressed NFκB-DNA binding activity after 48 h. The NFκB-DNA binding activity after HT was significantly (P < 0.001) reduced at all time points studied, and the maximum inhibition is observed at 48 h.

A: EMSA analysis showing a prolonged inhibition of NFκB-DNA binding activity upon exposure to HT. Cells were incubated at 37°C or exposed to 43°C for 60 min. After HT, cells were incubated in a CO2/air incubator for additional 10, 30 min, 1, 3, 6, 12, 24, and 48 h. B: Densitometric analysis showing the levels of persistent inhibition of NFκB-DNA binding activity in HT cells.
HT-Induced Constitutive Level of IκBα
To determine whether HT-induced inhibition of NFκB involves the induction of IκBα, cytoplasmic extracts obtained from the cells exposed to 37°C (Inc), 37°C (WB), 43°C for 15, 30, 45, and 60 min were subjected to immunoblotting analysis to determine the constitutive levels of IκBα. Compared to the controls, IκBα levels increased with increasing time period on 43°C, showing the maximum level in the cells that were exposed to 43°C for 1 h (Fig. 3A). This indicates the dose-dependent increase of IκBα in the cytoplasm (Fig. 3B). These results correlated very well with the results of the NFκB inhibition determined by the EMSA analysis, where the cells exposed to 43°C for 1 h showed maximum inhibition of NFκB-DNA binding activity.

A: Western blot analysis showing the constitutive levels of IκBα in cells incubated at 37°C and 43°C for 10, 30, and 60 min. After HT, the cells were incubated in a CO2/air incubator for additional 4 h. α-Tubulin expression was determined to validate equal sample loading. B: Densitometric analysis of HT-regulated IκBα expression. Quantitation was performed using Image Quant 1D gel analysis (Amersham Biosciences, Arlington Heights, IL). [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
HT-Induced Apoptosis and Reduced Colony Forming Capacity
To determine the HT-induced apoptosis, the MCF-7 cells exposed to 43°C for 1 h were stained with Annexin V-FITC and analyzed by FACS. Compared to control (37°C), HT (43°C for 1 h) significantly (P < 0.001) induced apoptosis (Fig. 4A). Like wise to determine the effect of HT on the colony-forming capacity, colony forming assay was performed after the cells were exposed to 43°C for 15, 30, 45 min, and 1 h. Cell survival after 43°C exposure was significantly (P < 0.001) reduced at all time points studied, and the maximum inhibition is observed at 1 h (Fig. 4B). The level of cell survival measured is clearly dependent on the time after exposure.

A: Increased expression of phosphatidyl serine indicating increased apoptotic cell death after exposure of cells to 43°C compared to cells incubated at 37°C. Expression of phosphatidyl serine was measured by staining the cells with Annexin V-FITC and analyzed by FACS. B: Colony forming assay showing inhibition of cell survival after 43°C exposure. Cells (300 cells/plate) seeded in a 60 mm culture plates were exposed to 43°C for 15, 30, or 60 min and incubated for 10 days. The colonies were stained with crystal violet and counted manually under low magnification.
HT-Induced Intracellular NO and Enhanced iNOS Expression
To determine the effect of HT on intracellular NO production, fluorescent intensity in the cells pretreated with fluorescent NO indicator DAF-2DA was analyzed after exposing the cells to 43°C for 1 h. SNP (1 mM) was used as the positive control. Compared to the control, HT (43°C for 1 h) significantly (P < 0.001) increased the NO production in MCF-7 cells (Fig. 5A). To determine whether HT inhibit the expression of iNOS, cytoplasmic extracts obtained from the cells exposed to 37°C, 43°C for 1 h, cells pretreated with GSNO, a NO donor and exposed to 37°C, cells pretreated with L-NAME, a NO inhibitor and exposed to 43°C, were subjected to immunoblotting analysis to determine the levels of iNOS. Cells pretreated with GSNO and exposed to 37°C showed an increased expression of iNOS. This serves as the positive control for the system. Compared to the control, HT significantly increased the iNOS expression in the MCF-7 cells (Fig. 5B). Consistently, in the cells treated with L-NAME and exposed to HT, the iNOS expression is markedly decreased. These results correlated very well with the results of the intracellular NO levels as determined by the DAF-2DA analysis, where the cells exposed to 43°C for 1 h showed significant induction of intracellular NO.

A: Histogram showing the levels of intracellular NO after exposing cells to 37°C (lane 1), HT (lane 2) and positive control (lane 3). The induced level of intracellular NO was examined by pre-incubating the cells with a fluorescent NO indicator DAF-2DA for 30 min before HT. The fluorescent intensity was measured by FACS analysis. SNP (1 mM) was used as positive control. B: Representative autoradiogram of immunoblotting demonstrating the induced level of iNOS expression after HT. Equal amounts of total protein extracts (50 µg) from the cells either incubated at 37°C or exposed to 43°C were analyzed for iNOS expression. To determine the specificity, cells were pre-incubated with a NO donor GSNO or a NO inhibitor L-NAME for 30 min before incubation at 37°C or exposed to 43°C, respectively. The experiment was repeated three times.
NO Mediated Inhibition of NFκB After HT
To determine the role of NO on the HT-induced inhibition of NFκB-DNA binding activity, cells were exposed to 37, 43°C for 1 hr or, pretreated with L-Name and exposed to 43°C for 1 h. After HT, the cells were incubated in a CO2/air incubator for additional 4 h. An autoradiogram showing the gel pattern is presented in Figure 6A. Densitometric analysis revealed a significant (P < 0.001) inhibition of NFκB-DNA binding activity, after the cells are exposed to 43°C for 15 min compared to the control. This HT-induced inhibition is significantly (P < 0.001) reverted (Fig. 6B) by pre-treating the cells with L-NAME suggesting that HT-induced inhibition of NFκB-DNA binding activity is NO mediated.

A: EMSA analysis showing inhibition of NO by L-NAME rescues the NFκB-DNA binding activity in MCF-7 cells exposed to 43°C for 1 h. B: Densitometric analysis of enhanced NFκB-DNA binding activity after inhibition of HT-induced NO by L-NAME. C: Phosphatidyl serine expression showing the inhibition of HT-induced apoptosis after either pre-treating the cells with NO inhibitor, L-NAME or after induced expression of NFκB. The induced expression of NFκB is achieved using transient transfection technique. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
HT-Induced Apoptosis Is Mediated by NO Regulated NFκB
To determine the role of NO on the HT-induced apoptosis, cells exposed to 37, 43°C for 1 h or, pretreated with L-NAME and exposed to 43°C for 1 h were stained with Annexin V-FITC and analyzed by FACS. Compared to control, HT (43°C for 1 h) significantly (P < 0.001) induced apoptosis (Fig. 6C). Consistently, in the cells pre-treated with L-NAME and exposed to HT, the Annexin V-FITC positive cells were significantly reduced. These results correlated very well with the results of the intracellular NO levels as determined by the DAF-2DA analysis, where the cells exposed to 43°C for 1 h showed significant induction of intracellular NO and with the expression results of iNOS levels as determined by the immunoblotting where the cells exposed to 43°C for 1 h showed significant inhibition of iNOS. Also these results correlate significantly with the results of the NO mediated inhibition of NFκB as determined by the EMSA. These date clearly indicated that HT-induced NO mediated inhibition of the NFκB-DNA binding activity enhance apoptosis in MCF-7 cells. To further substantiate our findings, cells transfected with pCNA3 containing full-length expression of NFκB p65 and p50 were stained with Annexin V-FITC and analyzed by FACS. Consistently, in the NFκB transfected cells, induction of apoptosis by HT (43°C for 1 h) were significantly (P < 0.001) reverted (Fig. 6C).
DISCUSSION
The anti-tumor activity of HT has been extensively characterized [Winter et al., 1985; Lindholm et al., 1987; Coleman et al., 1988; Herman et al., 1988, 1989; Petrovich et al., 1988; Shimm et al., 1988; Szmigielski et al., 1988; Valdagni et al., 1988; Overgaard, 1989; Overgaard et al., 1995; Vernon et al., 1996; van der Zee et al., 2000; Jones et al., 2002]. The importance of HT in cancer treatment was further supported by numerous clinical trials that clearly demonstrated the usefulness of local HT in improving local control [Lindholm et al., 1987; Valdagni et al., 1988; Overgaard et al., 1995; Vernon et al., 1996] and cell killing [van der Zee et al., 2000]. Furthermore, these studies demonstrated that HT induced local control involved cell killing and that, in certain cell types it involved apoptotic mechanism [Arends, 2000; Brade et al., 2003; Ma et al., 2004; Klostergaard et al., 2006]. The detailed mechanisms of heat-induced cytotoxicity or apoptosis remain to be elucidated. Several signaling proteins and transcription factors including NFκB [Mattson et al., 2004; Pajonk et al., 2005] may play important role in this response. NFκB has been consistently demonstrated to be a critical survival protein for many systems [reviewed in Piva et al., 2006], by brokering a myriad of downstream pro-survival and anti-apoptotic signals. In contrast, NO mediated stress has been causally linked to HT-induced apoptosis [Cui et al., 2006]. The results of the present study in mammary gland-derived estrogen-responsive MCF-7 cell line indicates that the inhibition of the anti-apoptotic transcription factor, NFκB by increased NO availability is one mechanism that mediates HT-induced apoptosis.
MCF-7 cells exposed to HT (43°C) downregulated NFκB-DNA binding activity in a dose-dependent manner and this induced inhibition is persistent (Figs. 1 and 2). After one-hour treatment and harvested at earlier time points (i.e., 10 and 30 min), the NFκB-DNA binding activity is dramatically reduced in the nuclear extract. This could be due to a cumulative effect of significant dissociation of DNA-bound NFκB subunits in the nucleus and complete shut-off of nuclear translocation from cytoplasm in response to 1 h of hyperthermia treatment. When the cells are incubated further for a longer time point, the pre-existing cytoplasmic NFκB, which might have survived as an intact protein through cellular protective measures against heat shock, now translocates into the nucleus and showed an increased level of DNA-binding activity compared to the earlier time point. This level is still markedly reduced compared to 37°C control. However, one should not underestimate the possibility of regaining a new synthesis by transcriptional activation of NFκB gene at prolonged period such as more than 96 h after 1 h treatment, which might reveal an increased NFκB binding as a cellular counter measure response and an attempt to repair the damage. This increased binding, however may not functionally viable to transactivate downstream gene in order to protect the cells from apoptotic death. As we have shown earlier with respect to other physical agents increased binding may not always results in transactivation of NFκB-dependent downstream genes [Natarajan et al., 2006]. Down-regulation of NFκB after HT has also been reported by other groups [Curry et al., 1999; Pritts et al., 2000; Mattson et al., 2004]. Pajonk and colleagues [Pajonk et al., 2005] attributed this to inhibition of 26S proteasome function, providing an alternative pathway for heat-induced NFκB inhibition through blocking IκBα degradation. Consistently, in the present study we observed a dose-dependent induction of the constitutive levels of IκBα after HT. In concordance with inhibition of constitutive NFκB activation, there is an increase in apoptosis and a dose-dependent decrease in cell survival. Ma and colleagues [Ma et al., 2004] demonstrated similar dose-dependent inhibition of cell survival and induced apoptotic cell death in MCF-7 cells after treating with 43°C for 30, 60, 90, and 120 min.
The relevance of NFκB to human malignancy is firstly based on the observation of enhanced DNA-binding activity in human clinical tumor samples, strengthened by its prognostic value [Mori et al., 1999; Lessard et al., 2003; Buchholz et al., 2005; Chen et al., 2005; Montagut et al., 2006]. Role of NFκB in cancer cell apoptosis has been extensively studied. Similarly, NFκB regulation in HT-induced apoptosis has also been shown previously [Kokura et al., 2003; Pajonk et al., 2005]. In addition to the dose-dependent inhibition of NFκB observed after HT, in the present study, we also observed that induced expression of NFκB in cells (transient transfection of NFκB sub units p65 and p50) significantly decreased the HT-induced apoptosis (Fig. 6C). Hence, a logical extension of our research was to delineate the mechanism of functional regulation of NFκB in HT-induced apoptosis.
Intriguingly, in this study we observed significantly induced levels of intracellular NO after HT (Fig. 5A). NO is a known regulator of both pro- and anti-apoptotic pathways [reviewed in detail by Dimmeler and Zeiher, 1997]. For instance, NO functions as an anti-apoptotic mediator by inhibiting caspase-3 activation via nitrosylation of procaspase-3 precursor [Mannick et al., 1999]. With an appropriate cellular stimulus (i.e., Fas ligation), denitrosylation of procaspase-3 occurs, thereby activating caspase-3 and initiating cellular apoptosis. Alternatively, NO may stimulate apoptosis by increasing cellular p53 levels (via an effect on proteosomal degradation) [Glockzin et al., 1999], by activating cytochrome c [Stamler et al., 2001], or perhaps by opening the mitochondrial permeability transition pore [Brookes et al., 2000]. Consistently, we observed an induced expression of iNOS after HT (Fig. 5B). NO produced by newly expressed iNOS might then act in concert with newly synthesized IκBα to inhibit the DNA-binding activity of NFκB and shut off the transcription of NFκB responsive anti-apoptotic and pro-survival genes. However, it is interesting to note that transcriptional activation of iNOS gene is mediated through four cis-acting NFκB elements residing in its promoter/enhancer region [Taylor et al., 1998]. This would counter argue against the observation of induced NO and inhibited NFκB after hyperthermia treatment. Although it is a general perception that the final outcome of the product in a cell depends on the genetic expression of the respective gene, with the recent advancement in the field of cellular functions clearly indicates that the regulation is equally contributed by the post transcriptional and/or post translational modifications. NOS induction and subsequent release of NO is regulated as a two step processes by multiple factors. First the protein expression of iNOS is mediated through transcriptional activation, but its enzyme activation depends on protein-protein interaction (Hsp-90 caveolin-1 etc.), site-specific phosphorylation, and post-translational modification of its prosthetic group. Next, generation of NO from iNOS is controlled by several factors including the bioavailability of its substrate, L-arginine, co-factors such as tetrahydrobopterin, and molecular oxygen. Hyperthermia treatment may alter one or more mediators in this regulation and induce NO without involving NFκB dependent promoter activation. Thus produced NO could down regulate NFκB by attenuates the phosphorylation of IkB. In addition, functional inactivation of NFκB by nitrosylation of NFκB subunits by increased production of NO after hyperthermia treatment cannot be ruled out. Ascertaining these mechanisms in NFκB mediated hyperthermia-induced apoptosis and the role of NO in this cascade of events is important and need further investigation.
To further substantiate our findings, we studied the NFκB-DNA binding activity and apoptosis after inhibiting the HT-induced intracellular NO with L-NAME, the NO inhibitor. Compared to the HT group, we observed an enhanced NFκB-DNA binding activity (Fig. 6) and reduced apoptosis (Fig. 6C) demonstrating for the first time, the role of HT-induced intracellular NO in inhibiting NFκB-DNA binding activity and promoting apoptosis. NFκB transcription is sensitive to oxidative and nitrosative stress [Marshall et al., 2000]. An oxidizing cytoplasmic environment is typically associated with NFκB activation, yet oxidation or nitrosation of the NFκB heterodimer (p50–p65) prevents DNA binding activity [Anderson et al., 1994; Matthews et al., 1996; Hirota et al., 1999]. These data suggest that NO/redox exerts control over NFκB at multiple loci within the signal transduction pathways that transmit inflammatory signals from the plasma membrane to the nucleus. p21ras, JNK kinase, and the p50 monomer (of p50-p65) have been identified as sites of S-nitrosylation that mediate the stimulation or inhibition of NFκB by NO [Matthews et al., 1996; Marshall et al., 2000; Marshall and Stamler, 2002]. Enhanced apoptosis after HT-induced inhibition of NFκB has been shown previously [Kokura et al., 2003; Pajonk et al., 2005], but this study is the first to link a physiological regulator of NFκB activity (i.e., NO bioactivity) with the NFκB-dependent control of HT-induced apoptosis.
It is noteworthy that induction of iNOS examined at 24 h after 44°C hyperthermia treatment for 15 min could increase the accumulation of wild type 53 in human glioblastoma cell line, A-172 [Matsumoto et al., 1999]. Although no direct link has been established, NO induced apoptosis in a p53-dependent manner has been documented in many cell types [Messmer et al., 1994; Messmer and Brune, 1996]. It can, therefore, be argued that hyperthermia-induced apoptosis through NO induction could involve p53 up regulation and its downstream effector protein p21. Alternatively, NO may stimulate apoptosis by increasing cellular p53 levels via an effect on proteosomal degradation) [Glockzin et al., 1999], by activating cytochrome c [Stamler et al., 2001], or by opening the mitochondrial permeability transition pore [Brookes et al., 2000]. However, on the same note studies showing: (i) no association between expression of iNOS mRNA and cell nuclear p53 protein in esophageal adenocarcinoma [Vaninetti et al., 2008], (ii) NO induction may directly targets p53 gene resulting in GC → AT mutations [Dijkstra et al., 1998; Martin et al., 2001; Staib et al., 2005] that may disrupt its apoptotic function, and (iii) NO induced DNA damage may lead to wild type p53 accumulation resulting in G1 growth arrest that allows the cells to repair the damage and block apoptosis [Li et al., 1996] in contrast to pro-apoptotic function discredit this argument. Anti-apoptotic or pro-apoptotic effect of induced endogenous NO in response to physical or chemical agents in cancer cells appear to depend on cell type, total NO levels, source of NO production, and concurrent production of superoxide [Fiscus, 2002; Fiscus et al., 2002].
In summary, in the present study using human adenocarcinoma (MCF-7) cells, we demonstrated the role of anti-apoptotic transcription factor, NFκB in HT-induced apoptosis. In addition, for the first time we demonstrated that the inhibition of the NFκB, is one mechanism by which NO mediates HT-induced apoptosis. The pursuit to dissect the molecular mechanism involved in HT-induced apoptosis is a potential fruitful strategy and, identification of a molecular target will greatly improve the efficacy of HT for the treatment of advanced and metastatic breast cancer.

