

Amplicon Resequencing Identified Parental Mosaicism for Approximately 10% of “de novo” SCN1A Mutations in Children with Dravet Syndrome
Communicated by Arupa Ganguly
Contract grant sponsors: Ministry of Science and Technology of China (No. 2012CB837600); National Natural Science Foundation of China (No. 81171221); Peking University Clinical Cooperation “985 Project” (PKU-2014-1-1 and PKU-2013-1-06); Beijing Municipal Science and Technology Commission (No. Z131100006813046).
ABSTRACT
The majority of children with Dravet syndrome (DS) are caused by de novo SCN1A mutations. To investigate the origin of the mutations, we developed and applied a new method that combined deep amplicon resequencing with a Bayesian model to detect and quantify allelic fractions with improved sensitivity. Of 174 SCN1A mutations in DS probands which were considered “de novo” by Sanger sequencing, we identified 15 cases (8.6%) of parental mosaicism. We identified another five cases of parental mosaicism that were also detectable by Sanger sequencing. Fraction of mutant alleles in the 20 cases of parental mosaicism ranged from 1.1% to 32.6%. Thirteen (65% of 20) mutations originated paternally and seven (35% of 20) maternally. Twelve (60% of 20) mosaic parents did not have any epileptic symptoms. Their mutant allelic fractions were significantly lower than those in mosaic parents with epileptic symptoms (P = 0.016). We identified mosaicism with varied allelic fractions in blood, saliva, urine, hair follicle, oral epithelium, and semen, demonstrating that postzygotic mutations could affect multiple somatic cells as well as germ cells. Our results suggest that more sensitive tools for detecting low-level mosaicism in parents of families with seemingly “de novo” mutations will allow for better informed genetic counseling.
Introduction
De novo mutations have been found to be the cause of many Mendelian disorders such as Dravet syndrome (DS) [Claes et al., 2001], Alport syndrome [Renieri et al., 1992], Marfan syndrome [Dietz et al., 1991], and tuberous sclerosis complex (TSC) [van Slegtenhorst et al., 1997; Veltman and Brunner, 2012]. They have also recently been found to be associated with complex diseases such as autism spectrum disorders, schizophrenia, and intellectual disability [Ku et al., 2013; Fromer et al., 2014]. De novo mutations are commonly believed to originate as postzygotic mutations in the germ cells of parents, which are transmitted to the offspring [Chandley, 1991; Veltman and Brunner, 2012; Ku et al., 2013; Ronemus et al., 2014]. Some postzygotic mutations in the parents may affect both their germ cells and somatic cells, a phenomenon commonly known as parental mosaicism [Biesecker and Spinner, 2013]. For some cases of parental mosaicism, the mutant alleles could be detected in the parental peripheral blood [Biesecker and Spinner, 2013; Poduri et al., 2013]. According to our literature review, for 110 non-cancer genetic disorders there have been one or more sporadic reports of causal mutations inherited from parental mosaicism [Bruttini et al., 2000; Jones et al., 2001; Depienne et al., 2006; Tekin et al., 2007]. In addition, parental mosaicism has been reported to be the origin of mutations in some families with two or more affected children carrying the same disease-causing mutation [Lee et al., 2011; Taioli et al., 2012].
DS is a severe epileptic syndrome (MIM# 607208) that occurs in the first year of life, characterized by fever-sensitive multiple seizures and refractory and psychomotor developmental delay after seizure onset [Roger et al., 1989]. Mutations in the gene encoding the alpha1 subunit of the sodium channel neuronal type I (SCN1A; HGNC# 10585, MIM# 182389) were identified in approximately 70% of DS probands in the Caucasian population [Escayg et al., 2000; Dravet and Oguni, 2013]. Most mutations in DS probands are considered de novo. Parental mosaicism has been reported in eight DS families in sporadic case reports using Sanger sequencing [Depienne et al., 2006; Shi et al., 2012] and in 13 DS families (7% out of 177) in one large study of a Caucasian cohort using Sanger sequencing and qPCR [Depienne et al., 2010]. In the latter study, 11 of the 13 mosaic parents had mutant allelic fractions over 5% and nine were detectable by Sanger sequencing [Depienne et al., 2010].
In current common practice, a mutation in a proband is considered “de novo” if Sanger sequencing detects the mutant alleles in the peripheral blood DNA of the proband but not in that of either of the parents [Claes et al., 2001; Eriksson et al., 2003; Hoischen et al., 2010]. However, because Sanger sequencing cannot detect mutant alleles with low allelic fraction (<5–10%) [Chen et al., 2013b], it is reasonable to suspect that parental mosaicism may be underdetected, especially when the allelic fraction is low. It was recently reported that four out of 100 “de novo” copy number variations (CNVs) in children with genomic disorders were in fact inherited from undetected parental mosaicism [Campbell et al., 2014]. Several other studies have also identified parental single-nucleotide mosaicism missed by Sanger sequencing using more sensitive technologies [Jones et al., 2001; Depienne et al., 2006, 2010]. These technologies include pyrosequencing [White et al., 2005], allele-specific quantitative PCR such as mismatch amplification mutation assay and high-resolution melting curve [Wittwer et al., 2003], TaqMAMA [Depienne et al., 2006]), digital PCR [Chen et al., 2013a; Campbell et al., 2014], denaturing HPLC (DHPLC) [Jones et al., 2001], mass spectrometry [Lee et al., 2012], restricted fragment length polymorphism [Selmer et al., 2009], single-strand conformation polymorphism/heteroduplex analysis [Orita et al., 1989], and the protein truncation test [Rohlin et al., 2009]. Recently, next-generation sequencing (NGS) technologies have been used to identify mosaicism [Rohlin et al., 2009] in cancer and 14 non-cancer diseases such as d-2-hydroxyglutaric aciduria [Nota et al., 2013], Alport syndrome [Artuso et al., 2011], and TSC [Qin et al., 2010]. Compared to other mosaic detection technologies, NGS-based methods have the advantage of being quantitative and having higher throughput. Single molecule molecular inversion probes can detect low-fraction alleles but specially designed probes are required [O'Roak et al., 2012; Hiatt et al., 2013]. In our research, we chose to use targeted PCR amplification, which is broadly applicable across the genome at low cost. We used ion torrent personal genome machine (PGM) semiconductor sequencing [Merriman et al., 2012; Chen et al., 2013b], which has the shortest run time and the lowest cost per run.
We investigated how many of the “de novo” mutations as determined by Sanger sequencing might in fact be undetected parental mosaicism in a cohort of 363 DS families in China. To detect and quantify mosaicism with improved sensitivity, we developed and validated a new protocol of amplicon resequencing using PGM and a hierarchical Bayesian model. We then validated the detected mosaicisms using pyrosequencing and digital droplet PCR. Finally, we studied the correlations between the mosaic mutant alleles and the phenotypes of the parents.
Materials and Methods
Patient Recruiting and Diagnoses
DS patients were recruited from the outpatient and inpatient child neurology units of Peking University First Hospital from 2005 till present [Sun et al., 2010]. The study was approved by the Ethics Committee of Peking University First Hospital and the Institutional Review Board at Peking University. Participants or their parents provided written informed consent before enrollment. We collected a large cohort of 363 Chinese DS probands and their families. All probands fulfilled the following diagnostic criteria [Baulac et al., 2004; Sun et al., 2010]: (1) seizure onset within 1 year of age (average age of onset 6 months) with the first event often being seizures induced by fever; (2) normal early development; (3) prolonged generalized or hemiclonic seizures, often triggered by fever; (4) multiple seizure types (myoclonic, focal, atypical absences) in addition to seizures triggered by fever after 1 year of age; (5) psychomotor slowing after 1 year of age, ataxia and pyramidal signs; (6) normal interictal electroencephalography in the first year of life followed by generalized, focal, or multifocal discharges; and (7) seizures that were pharmaco resistant. We described the detailed clinical phenotypes of 138 of the probands in a previous study [Xu et al., 2014], among whom 63 were screened for mutations in SCN1A [Sun et al., 2010]. The remaining probands were not previously studied or reported.
DNA Isolation and SCN1A Mutation Screen
Genomic DNA from peripheral blood lymphocytes was extracted using a salting-out procedure [Miller et al., 1988]. Genomic DNA from 20–30 hair follicles, buccal mucosa, saliva, and urine was isolated with the TIANamp Micro DNA kit (DP316; Tiangen Biotech, Beijing Co., Ltd.) following manufacturer's instructions. Genomic DNA from semen was isolated with the TIANamp Genomic DNA kit (DP304, Tiangen Biotech, Beijing Co., Ltd.) following manufacturer's instructions.
The 26 exons of SCN1A (NM_001165963.1) were amplified by PCR and analyzed using Sanger sequencing. Primers for the amplicons were listed in Supp. Table S1. CNVs were determined by multiplex ligation-dependent probe amplification (MLPA) as previously described [Sun et al., 2010]; the primers were listed in Supp. Table S2. When a nonsense or nonsynonymous mutation, frameshift insertion or deletion, or large insertion or deletion was detected in SCN1A in a proband's peripheral blood, his/her parents were screened for the same mutation using PCR Sanger sequencing.
For mutations determined by Sanger sequencing to be “de novo,” we subjected the parents’ DNA to a new protocol we developed to detect and quantify mosaicism using amplicon resequencing by PGM followed by a Bayesian model. The protocol, which we call PGM amplicon sequencing of mosaicism (PASM), was summarized in Supp. Figure S1 and described in the following sections. Details such as parameters are included in the Supporting Information.
Amplification and PGM Sequencing
Targeted PCR amplification was used to capture the genomic region around a mutation. To ensure amplification specificity, we designed two-stage nested PCR primers for mutations in genomic regions (200 bp upstream and 200 bp downstream around the mutation) that have a paralogous sequence elsewhere on the genome (i.e., ≥ 95% sequence identity over 200 bp determined by UCSC BLAT at http://genome.ucsc.edu/cgi-bin/hgBlat). Stage-1 amplification captured 1,000-nucleotide long amplicons (see primers in Supp. Table S3–1) to avoid nonspecific amplification, and Stage-2 amplification used these amplicons as templates and generated 200- to 400-nucleotide long products containing fused Ion Xpress barcode sets at the sequence termini (see original primers and fusion primers in Supp. Tables S3–2 and S3–3). Primer designs were described in the Supporting Information. The mutation must be more than 45 bases away from both ends of each amplicon (approximately falling within the 100–200 nucleotides, depending on the exact template and sequencing kits) because the ends were prone to sequencing errors.
Isolated and purified genomic DNA was amplified in a Takara Ex-Taq DNA polymerase (DDR100B, Takara) kit following the recommended instructions as previously described [Tajima et al., 2001] (Supporting Information). Modifications of the annealing temperatures were made for each primer pair to increase specificity (Supp. Table S3–3). Amplicons in each round were extracted with the Qiagen QIAquick® gel extraction kit (Cat. No. 28706; Qiagen, Hilden, Germany) after agarose gel electrophoresis (AGE) (G-10, lot no. 111910; Biowest). The AGE process served as size selection for amplicon uniqueness.
Library preparation for PGM followed the manufacturer's standard protocol (Supporting Information) with the following modifications to increase the quality of sequencing: all sets of purified barcoded amplicons were pooled to the same DNA molecule number relative to their molecular weight (in proportion to the amplicon length) and their original DNA concentration measured by Invitrogen's Qubit® High Sensitivity Assay (Cat. no. Q32852; Invitrogen by Life Technologies) on a Qubit 2.0 fluorometer. Longer amplicons (≧200 bp in a 200-bp library or ≧400 bp in a 400-bp library) were doubled, aiming to balance the amplicon abundance for emulsion PCR. Pooled amplicons were end repaired, ligated to ion-specific sequencing adaptors, and enriched in an additional 6-cycle PCR amplification following the manufacturer's recommendations (Ion Torrent by Life Technologies, Supporting Information).
Emulsion PCR and semiconductor sequencing on PGM were carried out following the manufacturer's instructions. Details about the protocol and the quality control steps were provided in the Supporting Information. We sequenced the amplicons to an average coverage of 32,830x.
Reads Preprocessing and Filtering
- (1) Base quality filter: Reads with low base quality usually had high sequencing error rates. In the prebase caller and base recalibration phase of Torrent Suite, we set “trim-qual-cutoff” to be 15, which would automatically trim bases with quality under 15 either before or after base quality recalibration.
- (2) Read depth filter: Loci whose coverage was less than 0.1% of the targeted average coverage were considered amplification failure and removed.
- (3) Strand bias filter: Unbalanced amplification of the forward and reverse strands (i.e., strand bias) was found to be associated with high sequencing error rates [McElroy et al., 2013]. We calculated the strand bias as follows. From all reads preprocessed and aligned by Torrent Suite and pileup files generated by SAMtools mpileup, we counted the total number of bases from the forward strand (
 ) and reverse strand (
) and reverse strand ( ). The number of reads mapped to a particular genomic nucleotide position in the forward and reverse strands were counted as
). The number of reads mapped to a particular genomic nucleotide position in the forward and reverse strands were counted as  and
and  , respectively.
, respectively. 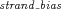 of a position is defined as
of a position is defined as

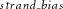 ,
, 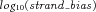 , is defined as
, is defined as

Genomic nucleotide positions whose absolute values of 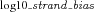 were ≥1 were considered having extreme strand bias and removed.
were ≥1 were considered having extreme strand bias and removed.
Finally, for reads that passed the above filters, at each genomic nucleotide position, Fisher's exact test was performed on the number of reference alleles on the forward strand, the number of mutant alleles on the forward strand, the number of reference alleles on the reverse strand, and the number of mutant alleles on the reverse strand from the pileup file, followed by Bonferroni correction. Positions with corrected P-value ≤ 0.05 were considered having strand bias and removed.
Hierarchical Bayesian Model to Calculate Allelic Fraction

 ,
,  is the likelihood of Bernoulli sampling, that is,
is the likelihood of Bernoulli sampling, that is, 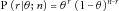 , which could be calculated when θ was given, and
, which could be calculated when θ was given, and  represents the summarized probability, which could be obtained by the iterative algorithm as described in the previous publication [Huang et al., 2014].
represents the summarized probability, which could be obtained by the iterative algorithm as described in the previous publication [Huang et al., 2014].Because the posterior distribution of θ was being calculated here instead of the integral, we modified our previous model by implementing a numeric method. We uniformly sampled θs within [0, 1] m times (m was set to 1,000 here), numerically calculated each  for r = 0 … n, multiplied each
for r = 0 … n, multiplied each  by its corresponding
by its corresponding  , and summed them up to get
, and summed them up to get  for each θ. We obtained
for each θ. We obtained for each of the m
for each of the m
 . For a better precision, a spline was fitted to
. For a better precision, a spline was fitted to  for interpolation. We then calculated the maximum-a-posteriori estimator, the 95% credible interval and the posterior mean using numeric integrations. For homopolymers and indels which were error-prone but did not get a base quality score from the PGM, we assigned an empirical base quality of 28, which was the mean base quality of 13,416,809 bases from the amplicon resequencing data of all the parents at all the genomic positions we tested. Ninety-five percent credible intervals were calculated by the same Bayesian model as described above. Details about the versions of software used in the pipeline were provided in Supp. Table S4.
for interpolation. We then calculated the maximum-a-posteriori estimator, the 95% credible interval and the posterior mean using numeric integrations. For homopolymers and indels which were error-prone but did not get a base quality score from the PGM, we assigned an empirical base quality of 28, which was the mean base quality of 13,416,809 bases from the amplicon resequencing data of all the parents at all the genomic positions we tested. Ninety-five percent credible intervals were calculated by the same Bayesian model as described above. Details about the versions of software used in the pipeline were provided in Supp. Table S4.
If the 95% credible intervals of the fraction of mutant allele (the same mutant allele as in the DS proband) detected by PASM in a parent were within the range [0.5%, 50%], the genomic nucleotide position was regarded as a mosaic site in that parent. Our protocol did not require a matched control sample, which was an important feature for studying non-cancer individuals who typically do not have matched control tissues. A modified criterion could be applied when a matched control sample was available: the 95% credible interval of the fraction of mutant allele in the matched control sample was calculated following the same procedure described above, and if it did not overlap with the 95% credible interval calculated from the sample of interest, the sample was regarded as having mosaicism at the genomic position tested.
Evaluation of the Accuracy of PASM Using a Serial Dilution Benchmarking Test
We evaluated the accuracy of PASM by a serial dilution benchmarking test. DNA from the blood sample of a proband with known heterozygous mutation in SCN1A (NM_001165963.1, c.1028+21T>C) was diluted with DNA from the blood of a normal control with homozygous reference alleles to obtain gradient samples with mutant allelic fractions of 0.5%, 1%, 2.5%, 10%, 25%, and 50%. PASM was applied to each gradient sample. Three technical replicates were performed to generate error bars.
We evaluated whether increasing PCR cycles would influence the measurement of the fractions of mutant alleles. DNA from a proband with heterozygous mutation in SCN1A (NM_001165963.1, c.4351C>A) was tested under PCR cycles ranging from 20 to 40 with 40 ng input template. Template inputs ranging from 20 to 80 ng with 40 cycles in a 50 μL PCR system were also tested.
Validation of the Parental Mosaicism Sites by Pyrosequencing and Digital PCR
We used two other quantitative experimental methods, pyrosequencing [Daskalos et al., 2011] and RainDrop digital PCR (RainDance Technologies, Lexington, MA), to validate the parental mosaicism sites detected by PASM. Amplification and detection primers for pyrosequencing were designed using Qiagen PyroMark Assay Design 2.0 software following standard procedures (Qiagen, Hilden, Germany). Genomic DNA amplification, post-PCR processing, and pyrosequencing experiments were conducted with the Qiagen PyroMark Q96 ID instrument (Qiagen, Hilden, Germany) using recommended materials and reagents (sequences provided in Supp. Table. S5). Data were processed and analyzed using the Qiagen PyroMark Q96 ID software to quantify the mosaic allelic fraction of each site measured. We have previously shown that technical replications of pyrosequencing had small variations [Huang et al., 2014], and thus only one pyrosequencing experiment was done for each sample. A previous study had reported that the detection limit of allelic fraction by pyrosequencing was approximately 1–5% [White et al., 2005], and thus signals below 1% were considered noise.
Because of this detection limit of pyrosequencing and because it could not properly distinguish insertions and deletions in regions with single nucleotide tandem repeat (also called homopolymer regions), we also performed RainDrop digital PCR (RainDance Technologies) for further validation of parental mosaicisms. Experiments were conducted following the manufacturers’ instructions [Chen et al., 2013a] and using customized TaqMan® assays (Supp. Table S6). Detailed experiment procedures were presented in the Supporting Information. PCR amplification program was setup following the recommended TaqMan® protocol with one modification: the heating and cooling rates of the thermocycler were adjusted to 0.6°C/s for better PCR amplification in millions of microdroplets. After PCR amplification, the sealed tube was put under a RainDance Sense chip for droplet reading. The raw data were processed with RainDrop Analyst V3.0 software. For each family, the signal of the heterozygous proband was used for signal compensation because they had strong signals in both channels representing wild-type and mutant alleles. The compensation procedure was performed following the manufacturer's user guide (RainDance Technologies). The fractions of the mutant alleles were calculated as the ratio between the number of the mutant targets and the sum of the numbers of the mutant and wild-type targets. Fractions of mutant alleles measured in heterozygous probands were adjusted to 50% for incomplete amplification assays, and 0% for negative controls; and a linear transformation was applied to fractions of mutant alleles in other samples measured with the same assay.
Prediction of the Functional Effect of Variants in SCN1A
To predict the functional effects of variants identified in SCN1A in the probands, we submitted the variants in variant call format to ANNOVAR (Version May 9, 2013) [Wang et al., 2010]. ANNOVAR predictions were made by SIFT [Ng and Henikoff, 2003], MutationAssessor [Reva et al., 2011], and Polyphen2 (Version 2.2.2) [Adzhubei et al., 2010] trained through human DIV database, following the recommended procedures at http://www.openbioinformatics.org/annovar/annovar_startup.html. GRCh37 was used as the reference genome. For comparison, common SNPs with population allele frequency over 1% were obtained from dbSNP version 137 as neutral variants and previously known causal variants for DS were collected from the SCN1A variant database at http://www.molgen.ua.ac.be/SCN1AMutations/ [Claes et al., 2009]. Protscale provided by the ExPASy server at http://web.expasy.org/protscale/ was used to calculate the possible changes to the protein three-dimensional structures induced by the amino acid alternations [Gasteiger et al., 2005].
Results
SCN1A Mutations in Probands and Families Affected by DS
We have established a large cohort of Chinese DS trio samples and screened for mutations in SCN1A in the peripheral blood DNA from 363 DS families using PCR Sanger sequencing and MLPA. We found that 255 (70.3% of 363) of the DS probands carried potentially damaging mutations in the SCN1A gene. Two (0.8% of 255) families admitted nonpaternity and were excluded from further studies. PCR Sanger sequencing for the same SCN1A mutations in the parents revealed that in 11 families (4.3% of 253) the mutations could be identified in the peripheral blood DNA from one of the parents as a heterozygous genotype, in five families (2.0% of 253) the mutations could be identified in one of the parents as mosaic, and in the remaining 237 families (93.7% of 253) the mutations were “de novo” (Fig. 1A). These 255 families had 223 different mutations, among which 167 (74.9% of 223) had never been previously reported in DS probands according to the SCN1A variant database. As shown in Figure 1B, 44.4% (99/223) of the mutations were missense mutations, 20.6% (46/223) were nonsense mutations, 5.8% (13/223) were small insertions, 16.1% (36/223) were small deletions, 9.4% (21/223) were splice site mutations, 0.5% (1/223) was a gene duplication, and 3.1% (7/202) were whole-gene deletions. The variant information was submitted to ClinVar (www.ncbi.nlm.nih.gov/clinvar/) and available with accessions SCV000221751-SCV000221973.

Validation of PASM by Serial Dilution Benchmarking Test
Results from the serial dilution benchmarking test showed that the fractions of mutant alleles measured by PASM were highly correlated with theoretical values (R2 = 0.94, Fig. 2A). This demonstrated that PASM was suitable for measuring mutant allelic fractions as low as 0.5%, which was beyond the detection limit of PCR Sanger sequencing, pyrosequencing, DHPLC, and traditional qPCR. The prebarcoded primers reduced library preparation costs by 90%. When the DNA samples were amplified in 20–40 cycles in the PCR system, the final measurement of allelic fraction by PASM was stable (Fig. 2B). A small amount of DNA (as little as 20 ng) was sufficient as the input templates to PASM (Fig. 2C).

Confirmation of the Five Cases of Parental Mosaicism Detected by Sanger Sequencing
For confirmation and comparison, the five cases of parental mosaicism detected by Sanger sequencing (DS001, DS002, DS003, DS004, and DS005; Fig. 3A) were subjected to PASM, pyrosequencing, and RainDrop digital PCR. Mutant allelic fractions in genomic DNA extracted from the peripheral blood of the mosaic parents were measured. All five cases of parental mosaicisms were confirmed by PASM (Fig. 3B). PASM quantified the mutant allelic fractions in the mosaic parents at 32.6%, 18.1%, 18.2%, 21.2%, and 13.3%, respectively, in positive controls at 48.30–56.07%, and in an unrelated normal control at 0.00–0.79% (Fig. 3B). Pyrosequencing was able to confirm four cases of parental mosaicism (DS002, DS003, DS004, and DS005; Supp. Fig. S2). The mutation in Family DS001 was a deletion in a homopolymer, which could not be accurately detected by pyrosequencing. Customized TaqMan® assays could distinguish between the mutant and reference alleles in two families (DS001 and DS004) and Raindrop digital PCR confirmed mosaicism in both of these cases (Supp. Fig. S3).

For the four families that could be quantified by pyrosequencing, the correlation between the fractions of the mutant alleles measured by pyrosequencing and PASM was high (R2 = 0.81, Fig. 3C). The correlation was lower for family DS004, and a close look at the results showed that pyrosequencing quantified some of the negative controls at 5–10% (Supp. Fig. S2), indicating possible detection limits of pyrosequencing. For the two families that could be quantified by Raindrop digital PCR (DS001 and DS004), the correlation between the fractions of the mutant alleles measured by Raindrop digital PCR and PASM in the mosaic parents, nonmosaic parents, and probands was very high (R2 = 0.95, Fig. 3C).
Detection of Parental Mosaicism in Families that Sanger Sequencing Considered Having “de novo” Mutations
We applied PASM to investigate how many of the “de novo” mutations in SCN1A might in fact be inherited from parent mosaicism undetected by Sanger sequencing. Out of the 237 DS families with paternity confirmed by clinical interview and where the probands’ SCN1A mutations were considered “de novo” by Sanger sequencing, 174 families still had remaining DNA samples from both parents available at the time of the PASM study. (DNA samples were available from both parents for all 237 families at the time of Sanger sequencing, but some samples were missing by the time of the PASM study.) We applied PASM to these 174 DS families. The probands in these families were found to carry “de novo” point mutations by Sanger sequencing and MLPA (Fig. 1A). As shown in Table 1, PASM discovered parental mosaicism in 15 (8.6%) of these families. The fractions of the mutant alleles ranged from 1.1% to 25.3%.
| Proband mutation information | Mosaic parent information | Mosaic related phenotype | Mosaic site information | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 95% credible interval | Validationc | ||||||||||||
| Family | Chromosome | Positiona | Nucleotide variationb | Amino acid variationb | Parent of origin | Reference allele | Mutant allele | Epileptic symptoms in parents | Fraction of mutant alleles by PASM (%) | Lower bound (%) | Upper bound (%) | Pyrosequencing (%) | Digital PCR (%) |
| DS017 | chr2 | 166848438 | c.5347G>A | p.A1783T | Father | C | T | Father, FS before 5 | 4.0 | 3.8 | 4.1 | 12 | 4.41 |
| DS027 | chr2 | 166915126 | c.337C>A | p.P113T | Father | G | T | Father, several FS at the early age | 25.3 | 22.3 | 28.5 | 43 | – |
| DS035 | chr2 | 166894440 | c.2792G>A | p.R931H | Father | C | T | Neither | 15.0 | 14.8 | 15.2 | 16 | 10.24 |
| DS094 | chr2 | 166848852 | c.4933C>T | p.R1645* | Father | G | A | Neither | 1.3 | 0.8 | 1.9 | 3 | 1.42 |
| DS101 | chr2 | 166848230 | c.5555T>C | p.M1852T | Father | A | G | Neither | 6.1 | 5.6 | 6.7 | 26 | 6.31 |
| DS104 | chr2 | 166904137 | c.1170+1G>T | – | Mother | G | T | Neither | 1.1 | 0.9 | 1.4 | 6 | – |
| DS117 | chr2 | 166895930 | c.2589+3A>T | – | Mother | T | A | Neither | 2.3 | 2.0 | 2.5 | – | – |
| DS125 | chr2 | 166868765 | c.3733C>T | p.R1245* | Father | G | A | Neither | 6.6 | 6.2 | 6.9 | 12 | 7.15 |
| DS128 | chr2 | 166868765 | c.3733C>T | p.R1245* | Mother | G | A | Neither | 13.2 | 12.4 | 14.1 | 19 | 13.02 |
| DS130 | chr2 | 166868772 | c.3726_3727insAT | p.D1243fsX1270 | Father | A | T | Neither | 3.3 | 2.8 | 3.9 | – | – |
| DS136 | chr2 | 166859043 | c.4223G>A | p.W1408* | Mother | C | T | Mother, undefined epilepsy | 9.2 | 8.5 | 9.9 | 22 | 11.71 |
| DS164 | chr2 | 166915194 | c.269T>C | p.F90S | Father | A | G | Father, FS at the early age | 8.6 | 7.9 | 9.4 | 15 | 9.32 |
| DS166 | chr2 | 166894396 | c.2836C>T | p.R946C | Father | G | A | Neither | 3.1 | 3.1 | 3.2 | 6 | 3.28 |
| DS188 | chr2 | 166894554 | c.2678T>A | p.L893* | Mother | A | T | Neither | 6.3 | 1.2 | 16.3 | 23 | – |
| DS206 | chr2 | 166901776 | c.1439_1442delCAGA | p.S481fs*488 | Father | G | A | Neither | 10.7 | 9.3 | 12.3 | – | – |
- a Position coordinates were based on the UCSC human reference genome version hg19.
- b Nucleotide and amino acid variations were based on RefSeq sequence NM_001165963.1. Numbering uses +1 as the A of the ATG translation initiation codon (codon 1).
- c Further details of the validation results from digital PCR and pyrosequencing are shown in Supp. Figures S4 and S5.
We applied pyrosequencing and RainDrop digital PCR for validation of the mosaicism. Specifically, pyrosequencing was applicable in 12 cases of parental mosaicism and validated all of them (Supp. Fig. S4). The R2 of the correlation between the fractions of the mutant alleles measured by pyrosequencing and PASM was 0.80. TaqMan® assays could be designed for nine cases of parental mosaicism and Raindrop digital PCR validated all of them (Supp. Fig. S5 and Table 1). The R2 of the correlation between the fractions of the mutant alleles measured by Raindrop digital PCR and PASM was 0.95. The lowest allelic fraction detected here was in family DS094 in which the mosaic father had mutant alleles at a low fraction of 1.4%, whereas the mother and a normal negative control had hardly any signal (fractions of mutant alleles under 10−5 which was in the range of noise, Supp. Fig. S5 and Table 1).
These 15 cases of parental mosaicism detected by PASM could account for 8.6% (15/174) of the mutations that were considered “de novo” by Sanger sequencing, which is the most common current practice. Taking these together with the five cases of parental mosaicism detected by Sanger Sequencing, we found a total of 20 cases of parental mosaicism. Our results suggest that parental mosaicism should be detected with more sensitive technologies and considered more seriously in genetic counseling.
Out of these 20 cases of parental mosaicism, 13 (65% of 20) originated from paternal mosaicism and seven (35% of 20) from maternal mosaicism. This was consistent with the predominantly paternal origin of de novo mutations previously reported in Mendelian disorders, complex diseases, and healthy individuals [Kong et al., 2012; Veltman and Brunner, 2012; Ronemus et al., 2014].
PASM detected mutant mosaicism in both the father and mother in Family DS082 (15.5% and 9.4%, respectively, for NM_001165963.1 c.4822G>T). Neither pyrosequencing nor Raindrop digital PCR was applicable at this genomic locus. We considered it a possible false positive by PASM and did not include it in Table 1. Family DS001 had mosaicism in the father detected by PASM and validated by Raindrop digital PCR. However, PASM also detected mosaicism in the mother (5.64%) but it was proven by Raindrop digital PCR to be a false positive (Fig. 3B and C and Supp. Fig. S3B and C). A close inspection revealed that the SCN1A mutations in families DS082 and DS001 were both located near homopolymers (both at the 3′ end of polyT). Homopolymers were known to be error-prone in PGM sequencing [Yeo et al., 2014] and might cause false positives by PASM.
Functional Prediction and Phenotype–Genotype Correlations of the Variants in SCN1A
The domain structure of the SCN1A protein sequence is shown in Figure 4A labeled with all the mutations in the coding regions identified in the DS families in which both parents’ DNA samples were available, including 11 families with inherited parental heterozygous mutations, 20 parental mosaic families (five detectable by Sanger sequencing and 15 by PASM), and 159 families with “de novo” mutations. Among all mutations, 90% (171 out of 190) were located in the coding region, including 31 frameshift, 46 nonsense, and 94 missense mutations, and the remaining 10% (19 out of 190) were located in splice sites. Among the missense single-nucleotide mutations, 98.9% (93 out of 94) were predicted to be deleterious by SIFT, MutationAssessor, or Polyphen2, and 93.6% (88 out of 94) were predicted to be deleterious by all three tools. The range of predicted functional effects of the mutations was similar to that of previously reported DS causal mutations in the SCN1A variant database and vastly different from that of common SNPs in dbSNP (Supp. Fig. S6, Column 3 vs. Columns 2 and 1). These results demonstrated that the mutations identified in this study were likely the causal mutations for the probands’ DS. The ranges of predicted functional effects were similar among the inherited, mosaic, and “de novo” mutations (Supp. Fig. S6, Columns 4–6).

We reviewed the clinical data of our DS cohort for epileptic phenotypes in parents including febrile seizures (FS), febrile seizures plus (FS+), or other epileptic syndromes. Epileptic phenotypes were present in 81.8% (nine out of 11) of the parents heterozygous at the site of the proband's mutation in SCN1A, 40.0% (eight out of 20) of the parents mosaic at the site of the proband's mutation in SCN1A, and 5.3% (17 out of 159 × 2 as we did not know the parent-of-origin) of either side of the parents of probands with “de novo” mutations in SCN1A (Fig. 4B). Thus, in general, parents with higher fractions of the mutant alleles had higher burden of epileptic phenotypes.
We further found that the fractions of mutant alleles in the mosaic parents with epileptic phenotypes were significantly higher than the fractions of mutant alleles in the mosaic parents without epileptic phenotypes (P = 0.016, single-tailed Wilcoxon rank sum test; Fig. 4C). Because almost all of the mosaic mutations were located in coil secondary structures within the SCN1A protein three-dimensional structure, we used Protscale to predict the level of changes in the local coil secondary structure caused by each mutation. Protscale predicted significantly higher effect on the coil secondary structure caused by mutations in mosaic parents with epileptic phenotypes than that by mutations in mosaic parents without epileptic phenotypes (P = 0.031, single-tailed Wilcoxon rank sum test; Fig. 4D).
Varied Fractions of the Mutant Alleles in Different Samples from the Same Parents
We obtained from two of the mosaic parents multiple samples in addition to peripheral blood including saliva, urine, hair follicles, and oral epithelium from the mother of DS004 (Fig. 5A) and saliva, urine, hair follicles, and semen from the father of DS001 (Fig. 5B). Analysis by PASM revealed that the mutant alleles could be detected in all samples at varied fractions (Fig. 5). The semen sample had higher mutant allele fraction than the other samples from the father of DS001 (Fig. 5B).

Discussion
Determining the origin of the mutations in probands is critical for genetic counseling. Our results suggest that parental mosaicism in DS is more common than previously thought. In this study, using a large cohort of DS probands, we found that 20 cases of SCN1A mutations that were not inherited from heterozygous mutations in parents were the consequence of parental mosaicism. We also found that as many as 75.0% (15 out of 20) of the parental mosaicism we detected with PASM could not be detected by Sanger sequencing. This implies that more sensitive technologies need to be implemented in the routine practice to determine the origin of mutations in probands. In addition to DS, sporadic cases of parental mosaicisms had been reported in over 110 other genetic diseases [Artuso et al., 2011; Nota et al., 2013]. We speculate that in many of these diseases, parental mosaicism may also play an important role that is underrecognized by current Sanger-based technologies.
Using PGM sequencing and a Bayesian genotyper, PASM was able to detect mutations with allelic fractions as low as 0.5%, a sensitivity significantly higher than Sanger sequencing and pyrosequencing [Hindson et al., 2011; Chen et al., 2013a]. Compared with the five cases of parental mosaicism identified by Sanger sequencing, PASM identified 15 more cases of parental mosaicism, an increase of 300%. In contrast, previous report indicated that qPCR only increased the number of detected parental mosaicism from Sanger sequencing by 33% [Depienne et al., 2010]. Compared to digital PCR technologies, NGS-based PASM did not depend on probes and thus could be applied to many more genomic loci. PASM required only trace amount of input DNA and was tolerant of a range of different PCR cycles for the sites we tested. Finally, PASM required only 20 h for sample preparation and sequencing, and the library construction cost of PASM was two-thirds of the original PGM protocol and one-third of the Illumina platforms, while providing the same amount of data. These features made PASM an attractive technology for detecting mosaicism. However, as exemplified in family DS001 (Fig. 3), the specificity of PASM needs to be improved for mutations near homopolymers.
PASM had detected three cases of parental mosaicism of small indels, including one small deletion that was detected by Sanger sequencing, and one small insertion and one small deletion that were undetected by Sanger sequencing. In the parents of probands that carried a whole gene deletion or duplication of SCN1A, MLPA did not detect any deletion or duplication. PASM was not applied on these parents because the current version of PASM could not detect mosaicism of large deletions or duplications. Previous study by Campbell et al. had found that four out of 100 “de novo” CNVs in children with genomic disorders were in fact inherited from undetected parental mosaicism [Campbell et al., 2014]. We expect that some of the “de novo” deletions and duplications in our cohort might also have been inherited from low-fraction parental mosaicism. In the future, we aim to develop a new version of PASM that could detect mosaicism of large deletions, duplications, and CNVs.
Using the current version of PASM, mutations in 159 families were considered “de novo”, that is, the mutations were not detected in either parent by PASM. However, several factors imply that some of these families might have low-fraction parental mosaicism missed by PASM. First, currently PASM could not detect mutations with allelic fractions lower than 0.5%; it is expected that low-fraction mosaicism could have been missed in some families. Second, the prevalence of epileptic phenotypes in the general population is approximately 3% [Hauser et al., 1993; Steinlein, 2002], whereas in our clinical data 5.3% of either side of the parents of DS probands with “de novo” mutations had epileptic phenotypes. Third, we found that “de novo” SCN1A mutations in probands whose parents had epileptic phenotypes had stronger impact on protein coil secondary structures (Supp. Fig. S7), a pattern similar to that of the mosaic mutations but weaker (Wilcoxon rank sum test, P = 0.069). Fourth, we observed that the PASM-measured fraction of mutant alleles of the heterozygous loci was slightly below 50% in the probands in four out of five families shown in Figure 3B and slightly below 50% for the benchmark samples shown in in Figure 2B and C. In Table 1, the PASM-measured fractions of mutant alleles were slightly lower than those measured by pyrosequencing and Raindrop digital PCR. A possible explanation for the skewed measurement was that the PCR conditions for amplification might be better for the reference alleles instead of the mutant alleles for these loci. However, there were insufficient data to make definitive conclusions. Finally, because we only confirmed paternity by clinical interview, not experimental testing, a small percentage of the families might have mispaternity, in which case any paternal mosaicism in the biological fathers would have been missed. Taken together, these factors implied that true percentage of parental mosaicism among DS families is likely to be even higher than our estimate.
We detected the mutant alleles at varied fractions in multiple samples from the same mosaic parents in both cases where samples were available. The mutations could be found in both somatic cells and germline cells, consistent with our previous report [Huang et al., 2014]. This implies that these mutations most likely originated in early embryonic development. We expect that postzygotic mutations that originated later in development would be more likely to have tissue specificity.
In previous literature, a total of 15 families had been reported to have two children diagnosed with DS [Escayg et al., 2000; Gennaro et al., 2006; Mancardi et al., 2006; Marini et al., 2007; Selmer et al., 2009; Depienne et al., 2010]. In our cohort, among the families with mutations inherited from a heterozygous parent, one family had two children (not twins) diagnosed with DS, carrying the same heterozygous mutation. Among the families with parental mosaicism detected, one family had a pair of monozygotic twins diagnosed with DS. Among the families with “de novo” mutations determined by PASM, there were three families each of which had two children diagnosed with DS. In two of these families, the affected were monozygotic twins. In the last family, the affected were non-twin siblings but no parental mosaicism was detected by PASM. Family DS002 (Fig. 3) had another child with FS or FS+ that died at a very young age. It was unknown whether she would fulfil the diagnostic criteria for DS if she had grown older and whether she carried the same mutation.
In this study, we focused on mosaic mutations that originated in the parents and were transmitted to offspring to cause DS. We did not investigate the possibility of mosaic mutations that originated in the probands themselves. There have been increasing reports of non-cancer diseases called by mosaicism in recent years [Biesecker and Spinner, 2013; Poduri et al., 2013; Freed et al., 2014], although no DS probands have yet been reported to be caused by mosaicism. The mutations in SCN1A that we identified in DS probands all appeared to be heterozygous germline mutations based on results from Sanger sequencing. We could not, however, rule out the possibility that more sensitive technologies may find some of the “de novo” mutations in probands to be mosaic, which would imply that a postzygotic mutation occurred very early in embryonic development in the probands. In addition, it is theoretically possible that more sensitive technologies may discover that some of the DS probands without mutations detected in SCN1A by Sanger sequencing may in fact have mosaic mutations in SCN1A with low allele fractions. In this case, additional sequencing is required to rule out the contribution of other germline mutations to confirm that the mosaic mutation is indeed the cause of DS.
With the rapid advances in sequencing technologies and digital PCR technologies, the detection of mosaicism will be further improved, which may bring new insights into the origin, transmission, and effect of mutations.
Acknowledgments
We thank the patients and their families for their participation and support.
Disclosure statement: The authors declare no conflict of interest.

