

Evaluation of kidney function throughout the heart failure trajectory – a position statement from the Heart Failure Association of the European Society of Cardiology
Abstract
Appropriate interpretation of changes in markers of kidney function is essential during the treatment of acute and chronic heart failure. Historically, kidney function was primarily assessed by serum creatinine and the calculation of estimated glomerular filtration rate. An increase in serum creatinine, also termed worsening renal function, commonly occurs in patients with heart failure, especially during acute heart failure episodes. Even though worsening renal function is associated with worse outcome on a population level, the interpretation of such changes within the appropriate clinical context helps to correctly assess risk and determine further treatment strategies. Additionally, it is becoming increasingly recognized that assessment of kidney function is more than just glomerular filtration rate alone. As such, a better evaluation of sodium and water handling by the renal tubules allows to determine the efficiency of loop diuretics (loop diuretic response and efficiency). Also, though neurohumoral blockers may induce modest deteriorations in glomerular filtration rate, their use is associated with improved long-term outcome. Therefore, a better understanding of the role of cardio–renal interactions in heart failure in symptom development, disease progression and prognosis is essential. Indeed, perhaps even misinterpretation of kidney function is a leading cause of not attaining decongestion in acute heart failure and insufficient dosing of guideline-directed medical therapy in general. This position paper of the Heart Failure Association Working Group on Cardio-Renal Dysfunction aims at improving insights into the interpretation of renal function assessment in the different heart failure states, with the goal of improving heart failure care.
Introduction
The interaction between the heart and kidney can often become deranged in heart failure.1 Not only do heart failure and chronic kidney disease (CKD) often co-exist and share common risk factors in their development, both heart and kidney disease can worsen each other's prognosis.2 Indeed, few organs in the body are such heavily intertwined in the proper execution of their physiologic function as the heart and the kidney.3 The preload of the heart directly depends on the sodium and water homeostasis regulated by the kidney. The kidney depends on adequate contraction and relaxation of the heart to have a sufficient trans-renal pressure gradient to maintain renal blood flow (RBF).4 Additionally, the majority of lifesaving guideline-directed medical therapies for heart failure have important direct effects on renal haemodynamics and solute handling. Although cardiologists are trained to evaluate the structure and function of the heart, less attention is directed at the evaluation of the kidney. Yet, the heart failure syndrome is characterized by haemodynamic, disease-related and treatment-induced alterations in kidney function.5 While some of these changes are merely a reflection of an appropriate renal response, others might indicate renal injury.6, 7 The misinterpretation of these alterations might result in inappropriate discontinuation of disease-modifying heart failure therapies, premature discontinuation of decongestive therapies, or ongoing renal injury.8-10 This position paper provides an overview about the evaluation and interpretation of kidney function throughout the heart failure trajectory.
Prognostic impact and terminology of renal function
Chronic kidney disease, defined as a glomerular filtration rate (GFR) < 60 mL/min/1.73 m2 or the presence of albuminuria (detailed definition in online supplementary Table S1) is present in 4.5% of the general population while it has a high prevalence in heart failure, affecting up to 50% of patients with either a preserved or reduced ejection fraction.11 The prognostic impact of any reduction in estimated GFR (eGFR) is well-established in heart failure.12-14 A large meta-analysis encompassing over one million patients with heart failure illustrated that the presence of CKD is associated with a doubling in the risk of all-cause mortality.11 Importantly, a reduced GFR is a stronger predictor of adverse outcome than a reduction in left ventricular ejection fraction in heart failure.15
In addition to the prognostic role of CKD, dynamic changes in renal function have also been recognized to portent a poor prognosis.16-18 However, such changes should always be interpreted in relation to the precise definition and the clinical context of the change in renal function.19 When discussing dynamic changes in renal function in heart failure, the terms ‘worsening of renal function’ (WRF) and/or ‘acute kidney injury’ (AKI) are often used.20 In addition, nephology literature has three sets of criteria to define AKI, namely the Risk, Injury, Failure, Loss, End-stage (RIFLE), Acute Kidney Injury Network (AKIN) and Kidney Disease: Improving Global Outcomes (KDIGO) criteria.21 Regrettably, the definitions used in medical literature vary greatly in terms of the degree of change (absolute vs. relative) and the used marker (creatinine, cystatin C, or eGFR).19 Table 1 gives an overview of the definitions of WRF and AKI in medical literature based upon biomarker changes. Importantly, on a heart failure population level, WRF or AKI, irrespective of the precise criteria used, are associated with a worse prognosis. However, as will be discussed later, WRF occurring during acute heart failure (AHF) with simultaneous favourable ongoing diuresis and improvement in heart failure status does not herald a more ominous prognosis (also termed pseudo-WRF).6, 22 A similar line of reason can be applied to the WRF occurring during initiation of neurohormonal antagonist therapy.23, 24 Particularly in the setting of heart failure with reduced ejection fraction (HFrEF), the treatment benefits of the neurohormonal blockers strongly outweigh the accompanying WRF, which itself is more an intra-renal haemodynamic reflection of neurohormonal blockade and not necessarily a signal of direct renal injury (pseudo-WRF).24 Yet, misinterpretation of these changes still results in inappropriate discontinuation of decongestive or neurohormonal blocker therapy in clinical practice.8, 25 Paradoxically, patients with baseline CKD (who are at higher risk for WRF) might actually benefit the most in absolute terms of treatment with neurohormonal blockers, as the presence of CKD is associated with a higher event rate.26-28
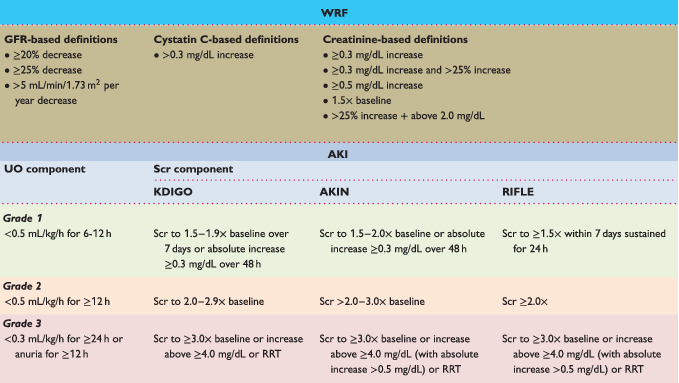 |
- AKI, acute kidney injury; AKIN, Acute Kidney Injury Network; GFR, glomerular filtration rate; KDIGO, Kidney Disease: Improving Global Outcomes; RIFLE, Risk, Injury, Failure, Loss, End-stage kidney disease; RRT, renal replacement therapy; Scr, serum creatinine; UO, urine output; WRF, worsening of renal function.
- To calculate serum creatinine in mg/dL to μmol/L multiply with 88.4.
- For AKI criteria, if urine output and serum creatinine stage do not correspond to the same stage, patients are classified in the worse stage.
- AKI criteria have a stage 1 (green line), stage 2 (orange line), stage 3 (red line).
- Adapted from Damman et al.19
Assessment of renal function
Renal function evaluation should be part of every encounter physicians have with heart failure patients as it helps (i) to better understand the underlying cardio-renal physiology, (ii) to improve initiation, adaptation or continuation of evidence-based heart failure therapies, (iii) to stratify patients at risk of adverse outcome, and (iv) to identify the presence of systemic diseases or the co-existence of independent renal disease. Importantly, it needs to be emphasized that renal function is more than just the assessment of glomerular filtration. As such, a wide variety of laboratory and imaging techniques are available to help the clinician to differentiate between functional and structural renal derangements in heart failure. Additionally, the interpretation of renal function also depends on the setting (AHF vs. chronic heart failure setting). Appropriate assessment of the presence of congestion in heart failure is essential in understanding renal function changes and has been discussed in a previous position statement.29 This section discusses the modalities to evaluate renal function in global, while the following sections give a contextual use in AHF and chronic heart failure.
Biomarkers
Biomarkers are naturally occurring molecules that can be rapidly, objectively and reproducibly quantified, preferably at a low cost, and offer insights into (patho)physiological processes.30 Although numerous cardio-renal biomarkers exist, to be of clinical utility, they need to be well studied in the clinical context of heart failure, and able to alter patient care on top of currently existing alternatives. As such, a prognostically relevant renal biomarker detecting AKI in nephrology cannot just be extrapolated to a situation of heart failure. Table 2 provides an overview of the potential prognostic, diagnostic and clinical utility of blood and urinary biomarkers studied in hearty failure.
| Marker | Prognostically relevant | Diagnostic for WRF | Therapeutically relevant |
|---|---|---|---|
| Blood biomarker | |||
| Glomerular function | |||
| Creatinine | ++ | ++ | ++ |
| Cystatin C | + | + | ? |
| Urea | +++ | ? | ++ |
| SuPAR | + | ? | ? |
| Pro-enkephalin | + | ? | ? |
| Tubular function | |||
| NGAL | ++ | - | ? |
| H-FABP | + | ? | ? |
| β2-microglubulin | + | ? | ? |
| Urine biomarker | |||
| Glomerular function and integrity | |||
| Creatinine | + | ? | + |
| Albumin | +++ | ? | ++ |
| Tubular function/injury | |||
| NGAL | ++ | + | ? |
| KIM-1 | + | + | ? |
| NAG | + | + | ? |
| Cystatin C | + | + | ? |
| β2-microglubulin | + | + | ? |
| NP′s | + | ? | ? |
| L/H-FABP | + | ? | ? |
| IGFBP7 | ? | + | ? |
| TIMP2 | ? | + | ? |
| Diuretic efficiency (natriuresis/mg diuretic) | +++ | +/? | ++ |
- H-FABP, heart type fatty acid binding protein; IGFBP7, insulin-like growth factor binding protein 7; KIM-1, kidney injury molecule 1; L-FABP, liver fatty acid binding protein; NAG, N-acetyl-β-D-glucosaminidase; NGAL, neutrophil gelatinase-associated lipocalin; NP, natriuretic peptide; SuPAR, soluble urokinase-like plasminogen activator receptor; TIMP2, tissue inhibitor of metalloproteinase-2; WRF, worsening of renal function.
Laboratory biomarkers of glomerular function
Aside from GFR, renal function also encompasses renal secretion and absorption. All these processes are altered in heart failure as they are influenced by intra- and extra-renal haemodynamics and neurohormonal activation.31-33 Plasma creatinine (enabling the calculation of eGFR) and urea are the only renal biomarkers that currently have a strong recommendation in heart failure guidelines (class I – level C).34 Mechanistically, GFR is the product of the number of functional nephrons and the single nephron GFR.35 The large number of nephrons at birth and the compensatory ability to increase single nephron GFR result in a large renal reserve capacity to maintain GFR and clearance.35 As such, GFR is reflective of the renal reserve, which partly explains its powerful predictive capacity of outcome in heart failure.
The gold standard of measuring GFR is by exogenous markers such as iothalamate or inulin as these molecules are freely filtered and largely without renal secretion or/and absorption. However, this is cumbersome and impractical for routine use in clinical practice.36 Therefore, endogenous filtration markers (creatinine or cystatin C) are measured that are fairly good estimates of gold-standard measured GFR. Serum creatinine is the most frequently used endogenous marker of glomerular filtration. Serum creatinine is a product of skeletal muscle creatine metabolism and is freely filtered by the glomerulus. However, it is also variably secreted by the tubules, hereby making it an imperfect marker of glomerular filtration.36 To overcome this imperfection, cystatin C has largely been studied based on the premise of only being filtered by the glomerulus and not secreted by the tubules.37 Cystatin C is a small molecule that is produced by all nucleated cells. Cystatin C is completely reabsorbed by the renal tubules, but broken down in the process, so that the reabsorption does not affect plasma levels. However, in states of tubular damage, the reabsorption is diminished and urinary cystatin C increases (indicating tubular injury, with urinary cystatin C not being informative of glomerular function). A study in heart failure patients suggests that cystatin C-based estimation of GFR is more precise that serum creatinine estimation.38 Understanding potential mechanisms of error is important when interpreting serum creatinine or cystatin C levels (online supplementary Table S2).39 For instance, cachexia, commonly occurring in heart failure, can result in a lower serum creatinine, hereby over-estimating glomerular filtration, if GFR is calculated based on creatinine solely. Alternatively, marked inflammation or obesity augment cystatin C for the same GFR. Additionally, the relationship between serum creatinine and eGFR is exponential.40 As a result, small changes in the lower range might indicate a significant impact on GFR, while larger changes in the higher range not necessarily mean a significant change in GFR.
Numerous formulas exist to estimate GFR based on serum creatinine, cystatin C, or a combination of both (online supplementary Table S2).39 While formulas exists using both serum creatinine and serum cystatin C, in most contexts of clinical practice, estimation based on solely serum creatinine probably suffices. Although the primary goal of these equations is to accurately approximate gold-standard measured GFR, depending on the objective, one formula might be preferred over another one.41 For instance, the Cockcroft–Gault formula has the worst accuracy in predicting gold-standard measured GFR, while it has been reported to outperform the simplified Modification of Diet in Renal Disease (sMDRD) formula in predicting outcome in heart failure41, 42 This is perhaps based on the fact that the Cockcroft–Gault formula incorporates weight, which is not included in the sMDRD or Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) formula. In chronic heart failure, the CKD-EPI formula has the best accuracy in predicting GFR.43, 44 As such, it is therefore commonly used in practice, also for instance for drug dose adjustments. However, it is important to mention that for drug registration and labelling, traditionally the Cockcroft–Gault formula has been used for GFR calculation and decisions how to adjust drug doses. As a result, CKD-EPI vs. Cockcroft–Gault might result in conflicting decisions when to adjust drug doses due to generation of different CKD classifications by different formulas.45, 46 For instance, the recent practical guide on the use of non-vitamin K antagonist oral anticoagulants (NOACs) recognizes that decisions on drug dosing for NOACs are best based on the Cockcroft–Gault formula, as this was used in landmark NOAC trials. Therefore, understanding the GFR-based equations is relevant in heart failure when dose adjustments are necessary with lower eGFRs.47 Perhaps for practical reasons it seems best to choose one formula to use in clinical practice. Finally, it is important to mention that all GFR estimation equations were primarily validated in stable patients, with stable creatinine values. By definition, they should track less well with true GFR during acute changes often occurring in AHF, where the fundamental assumption of steady state equilibrium of the filtration marker is violated. Indeed, a more holistic approach in AHF incorporating metabolic consequences (e.g. hyperkalaemia, acidosis, etc.) and diuresis and natriuresis might be more informative to get a global idea of kidney function.29, 48
Guidelines also advise the measurement of serum urea, which is not only related to glomerular filtration but also to tubular urea reabsorption and thus neurohormonal activation in heart failure.49 Proximal nephron sodium and water reabsorption results in solvent drag mediated more distal urea reabsorption. In addition, collecting ducts reabsorb urea under situations of vasopressin stimulation.50 Therefore, it is not surprising that urea is a powerful predictor of outcome. Other plasma biomarkers have been investigated to give information about glomerular function, but their precise role in heart failure management remains undefined51 (Table 2).
Laboratory biomarkers of tubular function
On a daily basis, the healthy kidneys filters 180 L of ultra-filtrate containing 1.5 kg of NaCl. However, significantly less than 1% of this NaCl and only a tiny fraction of other solutes are excreted into the urine, which illustrates that small derangements in tubular function might have a substantial impact on volume and electrolyte homeostasis.4 As the renal tubules consume the most oxygen in the kidney, they are sensitive to hypoxia, which is often present in heart failure when both renal arterial and venous flow are impeded. It is however very difficult to calculate or determine tubular function, especially when considering the different functions over the entire length of the tubules. Currently, there is no consensus on how to assess tubular function, resulting in a large number of investigated biomarkers30 (Table 2). Most of these markers can be found in the urine as they are produced or leak out of the tubular cells, while some can also be found in the plasma and are sometimes (partly) filtered or secreted and appear in urine as well.
The large majority of plasma tubular injury biomarkers have been investigated in the research setting, and many of these assays are not clinically available for bedside use. The most extensively studied plasma tubular injury biomarker is neutrophil gelatinase-associated lipocalin (NGAL).52 NGAL is freely filtered by the glomerulus and its plasma levels therefore reflect GFR to some extent. While plasma NGAL is related to inflammation and infection, urinary NGAL is thought to predominantly result from tubular production and secretion. Data on the exact relation between urinary and plasma NGAL are scarce, but both seem to increase in a state of AKI.53, 54 However, in the AKINESIS (Acute Kidney Injury N-gal Evaluation of Symptomatic heart failure Study) trial, plasma NGAL was not superior to plasma creatinine in predicting WRF or adverse in hospital outcome in patients with AHF.55 Other serum markers such as serum β2-microglubin and fatty acid binding proteins have been studied in smaller studies, also suggesting a relation with clinical outcome. At the moment, the current consensus is that clinical use of these biomarkers offers no incremental value on top of GFR and N-terminal pro-B-type natriuretic peptide (NT-proBNP) to guide clinical practice in heart failure.30
Urinary biomarkers, volume and composition
Obviously, as one of the main tasks of the renal tubules is to regulate sodium and volume status, which is particularly hampered in heart failure, a more precise assessment of the tubular function might the evaluation of the urine itself.3 Urine is easily sampled and readily available in clinical practice. Due to its direct relation to the nephron, it is exquisitely useful for the evaluation of renal function. Additionally, urine evaluation has been proposed as an earlier indicator of disease, as the kinetics of changes in urinary markers occur earlier than changes in serum markers such as creatinine, which often lags significantly.56 Numerous biomarkers can be measured in the urine including markers of glomerular function (e.g. urinary creatinine), glomerular integrity and podocyte function (e.g. albuminuria) and urinary markers of tubular function and injury (e.g. urinary tubular injury markers, urinary sediment analysis, urinary electrolytes).
Most importantly, urine electrolyte concentrations and urinary volume can be used as a functional test to determine the tubular function, which might be of particular interest in heart failure.57-60 A proper standardization of urinary collection (e.g. early freezing), and incorporation of baseline kidney function and hydration status, might be important especially when spot samples are used.
Evaluation of diuretic response (also termed diuretic efficiency) has gained a lot of interest lately.22, 60-67 Indeed, heart failure is characterized by a very early loss in natriuretic responsiveness, which contributes to development of congestion.1 Numerous studies in AHF have suggested that a good diuretic response is associated with better outcome.22, 60-66 Further details on diuretic response will be discussed in the section on renal function in AHF. Evaluation of glomerular function via 24 h urine collection with measurement of creatinine clearance is seldom used in clinical practice. Importantly, aside of the potential problem of incomplete collections, one needs to recognize that plasma creatinine concentrations change slowly in non-steady state conditions, leading to errors in GFR estimations. As such, 24 h urinary collection-based GFR assessments are only good reflections of the actual GFR in chronic heart failure patients with stable renal function. In case of uncertainty about GFR estimation based on formulas, creatinine clearance could be considered (based on the premise to be in a steady state situation).
Glomerular integrity is commonly evaluated by measuring albuminuria. In diabetes and CKD, high intra-glomerular pressures are thought to result in damage to the glomerular membrane and induce podocyte dysfunction, resulting in proteinuria and albuminuria.68-70 However, it is questionable that high intra-glomerular pressures are always present in heart failure, given the low renal perfusion pressure and flow, unlike the situations of diabetes, hypertension, or CKD.24 However, albuminuria is common in heart failure, with one-third having microalbuminuria and 10% having macroalbuminuria and its presence is linked to worse prognosis.71, 72 Although albuminuria might be a good target for renin–angiotensin–aldosterone system (RAAS) blockade therapy in diabetes and CKD where the reduction of albuminuria by these agents have been associated with improved outcome, there is currently no evidence supporting similarity in heart failure.73
Next to evaluation of glomerular function and integrity, urinary tubular biomarkers are commonly used to evaluate the potential presence of AKI (Table 1).
Most evidence originates from the nephrology literature were significantly elevated biomarkers such as urinary cystatin C, NGAL, kidney injury molecule 1, N-acetyl-β-D-glucosaminidase correlates well with proven injury on histopathologic examination of kidney biopsies.74 Other potential valuable ways to detect true AKI, such as urinary sediment analysis with the evaluation of the presence of muddy brown casts, granular casts and tubular epithelial cells remain unexplored in heart failure despite having a well-established role in nephrology.74
Opposed to AKI, there are no histologic studies linking tubular injury in patients with AHF presenting with WRF. In addition, most AHF patients who develop WRF based on serum creatinine/cystatin C or GFR criteria (Table 1) probably do not have true AKI (a situation called pseudo-WRF; see following sections). Indeed, the large majority of heart failure patients presenting with AHF and WRF do not have an increase in NGAL for instance. As such, it is not surprising that the magnitude of the elevation of these tubular ‘injury’ biomarkers is far less than in true AKI.6, 19, 60 Furthermore, elevated urine tubular ‘injury’ markers fail to identify heart failure patients at risk for poorer outcome or less diuretic responsiveness.6 Therefore, the adoption of these urinary tubular injury markers in heart failure is limited.
Renal imaging
Renal ultrasonography allows to measure kidney size (and abnormalities), which could be valuable given the high prevalence of CKD in heart failure. A sudden decline in renal function warrants imaging to rule out a urinary tract obstruction. Furthermore, renal artery evaluation should be considered in specific situations, such as severe decline in eGFR following initiation of a RAAS blocker.
It is well established both in acute and chronic heart failure that an increase in central venous pressures has a more pronounced impact on GFR than a decrease in cardiac output.75, 76 Indeed, enhanced renal venous pressures result in reduced RBF, with the latter being a significant determinant of GFR. Detailed echocardiography allows to assess cardiac filling pressures non-invasively.77, 78 However, in the process towards developing haemodynamic congestion, metrics of renal venous flow might become disrupted before metrics indicative of cardiac filling pressures (e.g. e′, E/e′, E/A ratio, systolic pulmonary artery pressure).79 Renal ultrasonography allows to assess such renal venous flow patterns, which can be measured bedside using an abdominal broad-band 2.5–5 MHz echo-probe. A continuous venous flow pattern is associated with low renal venous pressures, while increased venous pressures are associated with a discontinuous renal venous flow signal.80 Examples of different possible measurements are illustrated in online supplementary Figure S1. Interestingly, a discontinuous renal venous flow in response to volume expansion is associated with a reduced diuretic response independent of the underlying GFR.79 Although additional confirmation studies are warranted, renal venous flow pattern assessment might help to guide decongestive therapy.
Baseline assessment
Heart failure guidelines suggest evaluating renal function (creatinine, urea and eGFR) as a routine work-up in every patient diagnosed with heart failure (class I – level C).34 However, registry data indicate that only a limited amount of patients actually receive an appropriate work-up, for instance only 13–29% of heart failure patients receive a laboratory assessment of potassium and renal function after initiation of a mineralocorticoid receptor antagonist (MRA).81-83 Therefore, more emphasis should be given to appropriate baseline evaluation of renal function. In addition, it is the opinion of the Cardio-Renal Working Group that GFR estimation should preferably be performed using the CKD-EPI formula and health care professionals should be aware of the limitation of creatinine-based GFR calculations (as expressed in online supplementary Table S2).43 These latter two recommendations also have a class IB recommendation in the CKD KDIGO guidelines.84 In selected patients in whom creatinine-based GFR estimation might be less accurate (e.g. cachexia), physicians could consider measuring cystatin C at least once and use GFR estimation equations based on cystatin C (with or without creatinine). Given the high prevalence of CKD and albuminuria in heart failure, baseline evaluation of proteinuria and albuminuria using a morning urine sample may be considered.84 Additionally, cardiologists should be aware that specific cardiomyopathies present with significant non-albumin proteinuria (e.g. amyloidosis, Anderson–Fabry disease, mitochondrial DNA disorders).85 Finally, urine sediment analysis should be considered in the work-up of specific systemic disorders that could present with both intrinsic cardiac and renal disease (e.g. sarcoidosis, systemic lupus erythematosus, systemic sclerosis, etc.). Aside from specific situations in which a sudden unexplained renal function decline is present, the role of renal imaging, especially renal venous flow patterns, is currently under investigation.
Renal function in acute heart failure
The relationship between renal function changes, typically assessed as changes in creatinine or eGFR, and AHF is complicated.86 Worsening and/or improvement of renal function occurs commonly in AHF, affecting about 30–50% of admitted heart failure patients during hospitalization.6, 16, 87 Despite the direct epidemiologic link between changes in renal function and outcome, a thorough knowledge on the pathophysiologic contributors of renal function changes during AHF is essential. Not all changes in eGFR are equal, and appropriate interpretation is essential to improve heart failure care.6-11
Pathophysiology and prognosis of renal function changes in acute heart failure
Timing
In the setting of AHF, WRF and improved renal function have been associated with similar haemodynamic derangements and poor prognosis.6-11 Importantly, both definitions are used to describe renal function changes during hospitalization of AHF, and do not take into account changes that might have occurred before hospitalization. The fact that these changes most often occur during the first 3 days after admission suggests they are the result of haemodynamic derangements already present before hospitalization as well as a reflection of the therapies administered during hospitalization.20 Later changes are most often the result of effective decongestion or initiation/up-titration of neurohumoral blockade, which are not associated with worse prognosis, while later changes in GFR due to concomitant conditions (i.e. infection/sepsis) or administered nephrotoxic agents are associated with worse prognosis. In addition, changes in renal function are also a reflection of the underlying renal reserve to overcome the insult of AHF. As such, improved renal function may represent the resolution of venous congestion-induced pre-admission WRF and thus reflects a vulnerable kidney.
Pathophysiology
Not surprisingly, the underlying baseline GFR is one of the most important predictors for WRF during AHF.88 Numerous haemodynamic factors contribute to the development of renal function changes in AHF (Figure 1). A reduced arterial renal perfusion pressure secondary to an insufficient cardiac output has traditionally been regarded as a major contributor to a reduced RBF and subsequent reduction in GFR.5 While preserving adequate perfusion pressure is important,89 it is well recognized that reduced cardiac output only plays a minor role in the development of WRF, even in patients with advanced low-output failure.89 Indeed, patients with heart failure with preserved ejection fraction (HFpEF) and AHF have an equally high prevalence of WRF.88 Furthermore, the majority of HFrEF patients with AHF actually present with elevated instead of reduced blood pressures.90 Importantly, the glomerulus is capable of preserving its GFR in the face of a moderate reduction in RBF by changing the tonus of the afferent and efferent glomerular arterioles, thereby altering the filtration fraction.3 Importantly, more contemporary data illustrated that elevated central venous pressure more strongly affects RBF, which is associated with an impairment in baseline GFR and the development of WRF in both acute and chronic heart failure.75, 76 However, acute systolic blood pressure changes in the face of venous congestion also play a role in the development of WRF, indicating that episodes of frank hypotension should be avoided during decongestion.89 It is therefore obvious that a poor right ventricular function has also been associated with WRF as it leads to increased venous congestion.91 Furthermore, an increase in intra-abdominal pressures (normal range 5–7 mmHg; elevated >8 mmHg) is present in up to 60% of patients with advanced heart failure admitted with AHF.92 Such an increase in abdominal pressure is related to higher serum creatinine values, and strategies that reduce intra-abdominal pressures, such as diuretics, paracentesis or ultrafiltration, have been shown to reduce serum creatinine values.93 In addition, other non-haemodynamic factors including RAAS activation, sympathetic nervous system activation, inflammation, endothelial dysfunction and oxidative stress, also contribute to WRF development.94

In addition, the hospital trajectory of the patient with AHF is characterized by numerous interventions, such as initiation and up-titration of RAAS blockers and aggressive decongestive therapies,95-98 which might also result in WRF, typically occurring later during the course of heart failure hospitalization. Importantly, this type of WRF (often labelled pseudo-WRF) is not associated with worse outcome if associated with good decongestion in comparison to patients not attaining decongestion.97, 98 Indeed, WRF as the result of effective decongestion and/or implementation of disease-modifying therapies like RAAS blockers do favourably affect outcome. However, at the same time, hospital admission can also be complicated with other co-existing illnesses such as acute infections, sepsis, blood loss, which can also result in the occurrence of WRF, also typically later during the course of hospitalization.99 Similarly, exposure to nephrotoxic agents as iodine contrast, certain antibiotics or non-steroidal anti-inflammatory drugs (NSAIDs) during an AHF admission could also results in WRF. However, these latter pathophysiologic processes are also associated with a worsening in the patient status and potentially the development of an oliguric state (true WRF or AKI). Therefore, is important to interpret changes in creatinine (or eGFR) into potential contextual factors (Figure 1). However, the complexity of all factors that may (or may not) influence renal function and/or serum creatinine alterations in AHF makes it very difficult to establish cause–effect relationships.
Interpretation of changes in glomerular function
The goals of therapy in patients presenting with congestion and volume overload consists of (i) achieving thorough decongestion without residual volume overload; (ii) ensuring adequate perfusion pressures to guarantee organ perfusion; and (iii) maintaining guideline-directed medical therapies as these medications may also increase diuretic response and improve long-term survival.95, 100
During the interpretation of renal function changes during AHF, one should also incorporate the response to diuretic therapy (an example of tubular function assessment).29 Numerous metrics exist to assess diuretic response, using the total volume of diuresis, natriuresis, net fluid loss, or weight changes.60, 62-64, 66, 101 An overview of the different metrics and potential proposed cut-offs can be found in online supplementary Table S3. More recently, increased interest is being placed on urinary spot sodium concentrations the first hours after diuretic administration in the setting of AHF. Hereby allowing the clinician to interpret diuretic response and generating the opportunity to intervene early if sodium concentration is low. In the face of congestion with volume overload, a spot urine sodium concentration of <50–70 mEq/L after 2 h, and/or an hourly urine output <100–150 mL during the first 6 h, generally identifies a patient with an insufficient diuretic response.41, 67 However, irrespective of the metric used, all data indicate that a good diuretic response is associated with a better prognosis. Indeed, during AHF, the main objective is to get rid of all excessive volume by means of adequate decongestion, which is perhaps better attained in a setting of good diuretic response.29 Haemoconcentration provides a surrogate for a relative reduction in plasma volume between two time points and it therefore does not per se provide an indication of the absolute plasma volume (which might be the target).102 To overcome this, an instantaneous estimation of plasma volume (Duarte's formula) was derived from the Strauss formula (plasma volume changes).103-106 However, changes in haematocrit are often small, and can also relate to bleeding, phlebotomy, splenic pooling of blood and postural changes. Additionally, there remain some controversies regarding the reliability of these estimates in AHF. Additionally, plasma volume driven decongestive therapies have not been rigorously and prospectively tested as a guide to treatment in heart failure.
Interestingly, patients who have the best diuretic response often develop WRF, but at the same time have the best prognosis (pseudo-WRF).6, 22 It is therefore clear that evaluation of the kidney during AHF should not only be done by assessing glomerular function changes (development of WRF), but also by interpreting the tubular response to diuretic therapy (diuretic response/efficiency), and the ability to eliminate residual congestion and the administered therapy. Additionally extreme changes in creatinine should trigger the physician to think about the development of true WRF, especially when this is accompanied by other metabolic alterations (e.g. hyperkalaemia, acidosis). This should allow to identify patients with development of true WRF, who might have underlying kidney injury, vs. patients who have pseudo-WRF. Interestingly, a recent post-hoc analysis of the ROSE-HF (Renal Optimization Strategy Evaluation in Heart Failure) trial indicated that during aggressive decongestion, a substantial proportion of patients (21%) developed WRF. However, there was no association with the numerous urinary kidney injury markers. Patients who experienced WRF and a slight increase in urinary kidney injury markers during decongestion actually had the best prognosis.6 Nevertheless, more prospective studies are necessary to determine the best approach on how to optimally differentiate pseudo-WRF from true WRF and guide further treatment.
Therapeutic implications of worsening of renal function in acute heart failure
Strategies and tips on decongestion in the face of WRF are reflected in Figure 2. As alluded to, the goal of renal function evaluation during AHF is to better understand the pathophysiology at hand, in order to improve initiation, continuation or adaptation of heart failure therapies to improve the patient's heart failure status. Importantly, an increase in plasma creatinine often prompts physicians to reduce decongestive therapy, based on the often false assumption that further decongestion might result in renal tubular damage or failure. However, during decongestive therapy, an increase in creatinine should not automatically stop further decongestive therapy, especially if congestion persists. Indeed, clinical outcomes are extremely poor if patients are discharged with ongoing congestion in the face of WRF.107, 108 However, this does not indicate that all patients need to attain WRF to be deemed decongested. In contrast, an improvement in creatinine may provide false reassurance that decongestion has been achieved. The same line of thought is true when up-titration of neurohormonal blockers is being limited by solely a minor increase in serum creatinine. Therefore, changes in creatinine/GFR should always be seen in the clinical context/status of the patient. In line with a previous position paper, the use of a multi-parameter-based evaluation of congestion pre-discharge, using clinical assessment at rest and during dynamic manoeuvres as well as biomarkers, supplemented with technical assessments according to local expertise, is probably the best contemporary strategy to detect residual congestion.29

This Cardio-Renal Dysfunction Study Group proposes a repetitive comprehensive evaluation of WRF during AHF as described in Figure 2. As discussed earlier, the occurrence of WRF should be analysed in its context (timing, potential causes) and its relation with functional status and diuretic response (online supplementary Table S3). In patients with a good diuretic response, efforts should be taken to attain complete decongestion, as residual congestion at discharge is one of the main predictors of readmission.107, 109 Adequate dosing and early assessment of diuretic response through sodium excretion and urinary volume evaluation with early up-titration of diuretic doses if needed are essential.29 Furthermore, continuation, up-titration and (re)initiation of RAAS blockers should be considered in all HFrEF patients. Data indicate that despite a reduction in blood pressure and the occurrence of WRF, diuretic efficiency actually improves with up-titration of RAAS blockers.95 Despite that most evidence (albeit often retrospective) suggests that aggressive decongestion is associated with a better outcome, it is well known that loop diuretics induce neurohormonal activation. It is unknown if different diuretic regimens which induce decongestion but associated with less neurohormonal activation are associated with better outcome. Future ongoing studies will address this topic.110 Equally, the recent PIONEER-HF (Comparison of Sacubitril/Valsartan Versus Enalapril on Effect on NT-proBNP in Patients Stabilized from an Acute Heart Failure Episode) trial indicates that sacubitril/valsartan can be safely initiated during AHF and is associated with a reduction in NT-proBNP and a reduction in readmissions for heart failure, though not formally powered for this latter secondary endpoint.111 Importantly, only when the aforementioned therapeutic algorithm is followed, one can speak of an inefficient diuretic response when decongestion is not achieved with conventional therapy.
However, if diuretic response is poor and/or functional status deteriorates in the face of WRF, other causes should be sought (Figure 2). First, potentially correctable causes such as urinary tract obstruction or increased intra-abdominal pressures due to ascites should be excluded. Overall renal reserve (kidney sizes, degree of proteinuria, urinary sediments) should be assessed, and diagnostic work-up for the possibility of primary kidney failure (e.g. glomerulonephritis) should be sought, especially with potential underlying causes that may impact treatment decisions (e.g. lupus). Next, assessment of the haemodynamic status is needed. In haemodynamic stable patients, the presence of WRF and poor diuretic response might identify patients who are diuretic-resistant.61 A stepped pharmacologic diuretic regimen such as performed in the CARRESS-HF (Cardiorenal Rescue Study in Acute Decompensated Heart Failure) trial was capable of inducing significant diuresis in patients with acute decompensated heart failure and concomitant WRF.112 As specified before, a stepped pharmacologic care strategy which also incorporates the early evaluation of diuretic response has been suggested by this working group.29 The 2016 European Society of Cardiology (ESC) heart failure guidelines also suggest to consider the use of vasodilators in haemodynamic stable patients (systolic blood pressure > 90 mmHg) with AHF (class IIa).34 In contrast, in the ROSE-HF trial, the addition of low-dose dopamine or nesiritide did not result in better decongestion or less WRF after 72 h on top of aggressive decongestive therapy (2.5-times home loop diuretic dose).113 Additionally more recent trials on the use of vasodilators in heart failure have failed to show a beneficial effect on outcome, despite efficacy in lower blood pressure and relative good patient tolerability/safety.114-116 In patients with refractory volume overload and AKI, renal replacement therapy by ultrafiltration should be considered as a bail-out therapy (class IIa).117, 118 On the other hand, in patients with signs and symptoms of hypoperfusion and hypotension in the face of poor diuretic response in AHF, guidelines urge the optimization of the haemodynamic status, suggesting the admission to a critical care unit with invasive monitoring (class IC).34 Only in this minority of patients the use of inotropes, vasopressors or temporary mechanical support, should be considered (class IIb).34
Renal function in chronic heart failure
Total GFR is the product of both the number of functioning nephrons multiplied by the single nephron GFR.35 Interpretation of acute changes in GFR in AHF often relate to a temporary modulation of single nephron GFR function due to haemodynamic alterations or direct action of medication. Rarely – in the case of true AKI – these changes will result in a loss of functioning nephrons and relate to harm. In chronic heart failure, changes in GFR over time relate to a progressive loss of functioning nephrons despite a compensatory increase in single nephron GFR. As such, in chronic heart failure, unprovoked changes in GFR over time are strongly related to outcome.16 Importantly, when the total amount of functioning nephrons decreases, the reserve of the residual nephrons (increasing single nephron GFR) to oppose the stress induced by heart failure will be limited, resulting to subsequent accelerated nephron loss.
Evolution of renal function in health and heart failure
With aging, total GFR declines over time, which is related to a loss of functioning nephrons despite an increase in single nephron GFR, from the moment a critical mass of less than half a million nephrons has been reached.35 In healthy individuals, the averaged decline in loss of eGFR is around 0.6–1 mL/min/1.73 m2 per year.119 Risk factors such as hypertension, diabetes, obesity, albuminuria, diuretics and others are associated with a risk for a faster decline in GFR slope (Figure 3). Limited data are available about the effect of heart failure on progression towards CKD and the evolution of the slope of GFR. An analysis of over 3.4 million patients without heart failure matched to 156 743 patients with heart failure indicated that heart failure patients had a 2.12 higher risk for progression towards CKD.120 Additionally, heart failure patients are 2.96 times more likely to manifest with a rapid decline in GFR slope (defined as >5 mL/min/1.73 m2 per year), which occurs in 22% of heart failure patients.120 In line with that, the GISSI-HF (Gruppo Italiano per lo Studio della Sopravvivenza nell'Insufficienza Cardica-Heart Failure) trial reported the average decline of the GFR slope for heart failure patients to be 2.57 mL/min/1.73 m2 per year121 (Figure 3). Importantly, heart failure itself remains independently associated with a more pronounced decline in eGFR over time, after adjustment for other well-known risk factors associated with progression towards CKD.121 Additionally, a more rapid decline in eGFR is associated with a higher risk for adverse events.122

Impact of guideline-directed medical therapies on renal function
Despite the observational data illustrating that heart failure is associated with a more pronounced decline in eGFR over time, there is actually a paucity of data indicating that pharmacologic therapies for heart failure might be able to reduce the slope of this decline. Nevertheless, the beneficial effects of RAAS inhibition and sodium–glucose co-transporter 2 (SGLT2) inhibition on the slope of GFR decline observed in patients with type 2 diabetes and CKD are often extrapolated to heart failure patients.70, 123-125 Yet, in terms of intra-glomerular haemodynamics these disease conditions are not similar. Diabetes and CKD are associated with intra-glomerular hypertension due to glomerular hyper-filtration, which increases single nephron GFR to a supra-physiological extent and results in an accentuated loss of individual glomeruli. Unloading of the glomerulus (reduction in intra-glomerular pressures) via efferent arteriolar vasodilatation by RAAS inhibition has been shown to beneficially influence the decline in GFR slope in diabetic patients and patients with CKD.70 As these patients often have aldosterone escape and residual increased intra-glomerular pressures despite RAAS inhibition, it is not surprising that SGLT2 inhibition has shown an incremental beneficial effect on GFR deterioration. Indeed, SGLT2 inhibition results in further unloading of the glomerulus by inducing afferent arteriolar vasoconstriction.126
Less information exists if heart failure patients also have intra-glomerular hypertension. Due to haemodynamic and neurohormonal alterations, RBF in heart failure is reduced, which will induce an auto-regulation response of the afferent arteriole (vasodilatation) and the efferent arteriole (vasoconstriction), aiming to maintain GFR.31, 32 Yet the net effect on intra-glomerular pressures is not always clear, especially if patients are treated with RAAS inhibitors. As the body aims to preserve GFR, the filtration fraction (which is the ratio of GFR/RBF) will be altered. Therapies that have an effect on auto-regulation such as RAAS inhibition will therefore reduce GFR33 (Figure 3). As such, a drop of eGFR up to 15–20% might be anticipated after the initiation of RAAS inhibition. However, this phenomenon is an intra-glomerular haemodynamic feature, which is most often transient when discontinuing RAAS inhibition and is not associated with a loss in functioning nephrons and therefore does not induce intrinsic renal damage. Additionally, the slope of renal progression is similar following initiation of RAAS inhibition after the initial drop. However, it is unknown if after longer follow-up, angiotensin-converting enzyme inhibitor (ACE-I)/angiotensin receptor blocker (ARB) use (without concomitant neprilysin inhibition) is associated with a less steep slope of eGFR decline (Figure 3). Importantly, renal dysfunction is one of the most frequent reported reasons for under-dosing of guideline-directed doses of RAAS inhibitors.8, 25 Understanding the benefits of guideline-directed medical therapy even in the face of a small reduction in eGFR in heart failure patients, especially in patients with co-existence of CKD, is therefore essential.
Renin–angiotensin–aldosterone and neprilysin inhibition
Post-hoc analysis of landmark ACE-I trials as well as real-world observational data in ambulatory symptomatic HFrEF patients have illustrated a beneficial effect of ACE-inhibition, also in patients with baseline CKD and in those who experienced a drop in eGFR after initiation of ACE-I.127, 128 In CONSENSUS (Cooperative North Scandinavian Enalapril Survival Study), 11% of patients assigned to enalapril experienced a doubling of serum creatinine.129 This occurred early in most and serum creatinine returned back to within 30% of baseline values in the majority of patients.130 Although 33% of patients in SOLVD (Studies of Left Ventricular Dysfunction) had an increase in serum creatinine of >0.5 mg/dL, the benefits on outcome were well maintained, even in patients with more advanced (pre-dialytic) CKD131 (Figure 3). Less data are available for ARBs, however a propensity adjusted analysis illustrates a similar benefit on outcome despite presence of CKD.132 Additionally, patients with hyponatraemia (a marker of neurohormonal activation) are more likely to develop WRF after the initiation of ACE-I/ARB.133 However, this perhaps identifies a more sicker patient population and does not imply a reduced benefit of these agents. Though the benefits on morbidity and mortality with ACE-I/ARB treatment are sustained despite an initial drop in eGFR, there are no specific data from heart failure trials that treatment with ACE-I/ARB also reduces the slope of GFR decline in comparison to patients assigned to placebo. It needs to be emphasized that the follow-up of landmark ACE-I and ARB trials was relatively short, which may preclude the detection of alterations in the slope of eGFR decline. Additionally, ACE-I and ARB have shown to be reno-protective in patients with CKD and diabetes, which still constitute a large subgroup in the heart failure population.
In contrast, it has been demonstrated that the combination of neprilysin inhibition on top of an ARB (sacubitril/valsartan) does reduce the slope of GFR decline in patients assigned to sacubitril/valsartan in comparison to enalapril.23 Furthermore, both observational and trial data indicate a higher likelihood of loop diuretic down-titration in patients treated with sacubitril/valsartan in comparison to ACE-I/ARB.134, 135 Nevertheless, it remains always important to consider loop diuretic down-titration in patients treated with RAAS antagonists as complications related to these agents (changes in serum creatinine, hypotension, diminished urine output) often result from inadequate intravascular perfusion and are often reversible with diuretic dose adjustments. The beneficial effects of sacubitril/valsartan on morbidity and mortality are also well maintained in patients with more advanced CKD. Additionally, the incidence of hyperkalaemia is also lower in the sacubitril/valsartan arm in comparison to the enalapril arm.23, 136 Both in the PARADIGM-HF (Prospective Comparison of ARNI with ACEI to Determine Impact on Global Mortality and Morbidity in Heart Failure) and PARAMOUNT (Prospective Comparison of ARNI with ARB on Management of Heart Failure with Preserved Ejection Fraction) trials, initiation of sacubitril/valsartan was associated with an increase in urinary albumin creatinine ratio, which was transient after discontinuation of sacubitril/valsartan.23, 137 Although an increase in urinary albumin is generally associated with a higher risk, this is not the case when it comes to initiation of sacubitril/valsartan.23 A potential explanation might be that natriuretic peptides can have a transient effect on mesangial cells (or podocytes), altering hydraulic conductivity of the glomerular filter, without causing irreversible glomerular damage.24
Further suppression of the RAAS axis using a MRA beneficially influences outcome in HFrEF patients as illustrated in the RALES (Randomized Aldactone Evaluation Study) and EMPHASIS-HF (Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study) trials.138, 139 A post-hoc analysis of the EMPHASIS-HF trial illustrated that the presence of an eGFR <60 mL/min/1.73 m2 did not influence the benefit on the primary endpoint of heart failure hospitalization and cardiovascular mortality.140 Similarly to ACE-I and ARB initiation, analysis from the EMPHASIS-HF trial indicated that MRA initiation causes an acute drop in GFR which is maintained throughout MRA administration, although the absolute drop is minor (adjusted mean difference of −1.40 mL/min/1.73 m2).140 Importantly, MRA trials have generally excluded patients with more advanced CKD (i.e. <30 mL/min/1.73 m2). In addition, assessment of potassium is warranted after initiation as real-world pick-up of MRAs following the publication of the RALES trials resulted in increased incidence of hyperkalaemia.141 New strategies with oral potassium binders (patiromer or zirconium cyclosilicate) might improve the prescription and dose of RAAS blockers and MRAs in patients with CKD by reducing the rate of hyperkalaemia,142 especially in euvolaemic patients with hyperkalaemia in which further up-titration of loop diuretics is not possible. The ongoing DIAMOND (Patiromer for the Management of Hyperkalaemia in Subjects Receiving RAASi Medications for the Treatment of Heart Failure, NCT03888066) trial will assess if treatment with patiromer is associated with a lower risk for cardiovascular death or cardiovascular hospitalization.
Beta-blockers
Beta-blockers significantly reduce mortality and morbidity in HFrEF patients. Contrary to RAAS inhibitors, beta-blockers do not cause an acute reduction in eGFR or alter the slope of eGFR decline over time.143 A post-hoc analysis from the MERIT-HF (Metoprolol CR/XL Randomized Intervention Trial in Congestive Heart Failure) trial across GFR strata (<45, 45–60, and >60 mL/min/1.73 m2) indicates that patients with the lowest GFR strata actually had the highest relative risk reduction effect of metoprolol.144 It is important to emphasize that patients with the lowest GFR also have the highest baseline event rate.2 Therefore, prescription of beta-blockers to patients with CKD has an even larger impact in absolute terms. Similar trends were found in a sub-analysis of the SENIORS (Study of Effects of Nebivolol Intervention on Outcomes and Rehospitalization in Seniors with Heart Failure) with nebivolol and CIBIS-II (Cardiac Insufficiency Bisoprolol Study II) trials with bisoprolol.145, 146 However, the same analysis from CIBIS-II also illustrates that beta-blocker discontinuation is the highest in patients in the poorest GFR strata.146
Other heart failure therapies
A sub-analysis from the SHIFT (Systolic Heart Failure Treatment with the If Inhibitor Ivabradine Trial) trial indicated that ivabradine is equally effective in reducing the primary endpoint of heart failure hospitalization and cardiovascular death in patients with or without renal dysfunction.147 Though a higher heart rate in patients included in SHIFT was associated with an increased risk for WRF, ivabradine itself did not alter the eGFR over time in comparison to the placebo group.
Patients with CKD KDIGO stage 4 and 5 have generally been excluded from cardiac resynchronization therapy (CRT) trials.148-152 However, a large propensity-matched analysis of almost 11 000 patients with CKD stage 3–5 indicated that CRT with defibrillation capability was associated with a better outcome in comparison to patients receiving an implantable cardioverter-defibrillator.153 Interestingly, data indicate that CRT was capable of even improving eGFR.154 Additionally, following CRT implant, reduction in the dose of loop diuretics is often feasible, which might also partially explain the beneficial effect of CRT on renal function.155 Indeed, despite having no randomized placebo controlled trial data on loop diuretic use, observational data indicate that chronic loop diuretic use is an independent predictor of accelerated renal function decline.156 Nevertheless, despite extensive covariate and propensity adjustment in these observational studies, residual bias cannot be excluded. The renal interactions of diuretics in the chronic setting are complex and involve numerous pathways. A detailed discussion falls beyond the scope of this manuscript, but have been reviewed recently.29
Therapies negatively influencing renal function
Several therapies should be avoided in heart failure as they may result in worsening of heart failure. Numerous mechanisms exist depending on the administered drug including direct myocardial toxicity, enhancing bleeding risk and enhancing risk for arrhythmias. A detailed description spans beyond the scope of this position paper, but has been reviewed previously.157
In addition to the aforementioned mechanisms of direct toxicity to the heart, some medications result in worsening of heart failure by negatively affecting glomerular function and/or through increased sodium avidity. NSAIDs enhance renal resistance at mainly the level of the afferent arterioles, hereby reducing RBF and GFR.158 Furthermore, they enhance the filtration fraction of the glomeruli, a process that results in proximal sodium retention on its own. Finally, a decrease in prostaglandins might also result in sodium avidity at the level of the thick ascending limb of the loop of Henle and enhanced free water retention at the level of the collecting ducts. As such, they increase the risk for heart failure decompensation and have a class III recommendation in heart failure.34 Surprisingly, AHF registries seldom recognize NSAID use as a precipitating factor for the development of AHF.159 Yet, real-world pharmacy record studies indicate that the use of NSAIDs in heart failure patients might be as high as 30%.160
In addition to NSAIDs, the use of an ARB or direct renin inhibitor on top of ACE-I is not recommended in HFrEF as it is associated with an increased risk for WRF and hyperkalaemia.34, 161 Furthermore, the use of thiazolidinediones is not recommended to treat diabetes in heart failure patients as they cause sodium retention (class III recommendation), through up-regulation of the eNaC channel in the distal nephron.31 Furthermore, some medications could potentially be detrimental due to their high sodium content. Although data on the importance of sodium restriction are controversial in heart failure, guidelines recommend the avoidance of high diets with a high salt content (>6 g), through stabilizing dietary sodium intake. Currently, the SODIUM-HF (Study of Dietary Intervention Under 100 mmol in Heart Failure, NCT02012179) is investigating if a more liberal use of sodium is linked to better prognosis. Online supplementary Table S4 illustrates several commonly used medications that have a high salt content and might worsen heart failure.
Therapeutic implications of worsening of renal function in chronic heart failure
Patients with heart failure benefit from regular follow-up and monitoring of biochemical parameters to ensure the safety and optimal dosing of medication and to detect complications or disease progression that may warrant a change in management plan. An approach endorsed by this working group to WRF in chronic heart failure is reflected in Figure 4. The ESC heart failure guidelines recommend at least an analysis of serum creatinine, urea, eGFR, sodium, potassium every 4 months if patients are on stable doses of RAAS inhibitors.34 In patients with more advanced CKD, KDIGO guidelines recommend laboratory follow-up every 3 months. During titration of RAAS inhibitors, more frequent laboratory analysis should be performed, with ESC guidelines recommending to check creatinine and potassium 5–7 days after up-titration of MRA and within 2 weeks after up-titration of ACE-I or ARB. It is important to emphasize that contemporary data illustrate that such frequent laboratory follow-ups are hardly performed in clinical practice.82, 83 Also, before changing the dose of neurohormonal blockers including MRAs, haemolysis with false elevate potassium needs to be excluded. To date, there are no data to support the use of novel plasma and urinary biomarkers, as reflected in Table 2, to guide treatment in heart failure. Although urinary sodium concentrations carry a strong predictive capacity in AHF, limited data are available about its value in spot urine samples in patients with chronic heart failure.57, 59, 60, 64 One recent study indicated that stable chronic heart failure patients with a low urinary sodium concentration are more likely to develop AHF, after correction for NT-proBNP and eGFR.162 Furthermore, urinary sodium concentration decreased significantly the week before the development of AHF.162 However, the clinical applicability of routine use of simple biomarkers such as urinary sodium to guide treatment (e.g. diuretics or salt restriction) remains to be determined.

Conclusion
Impaired kidney function is one of the strongest predictors of outcome in heart failure. Yet interpretation of changes in kidney function in the appropriate clinical context is essential for optimal therapy in both chronic and acute heart failure. An approach to also assess tubular function (diuretic response) beyond assessments of GFR, especially during AHF, ensures appropriate assessment of renal function and restoration of salt and water balance. Equally, in chronic heart failure, correct interpretation of changes in serum creatinine during titration of RAAS inhibitors helps to implement delivery of the most optimal treatment options.
Conflict of interest: W.M. has received research grants from Novartis, Vifor, Medtronic, Biotronik, Abbott and Boston Scientific. J.M.T. reports grants and personal fees from Sequana Medical, BMS, Boehringer Ingelheim, Sanofi, FIRE1; personal fees from Astra Zeneca, Novartis, Cardionomic, Bayer, MagentaMed, Reprieve Medical, W.L. Gore; grants from 3ive labs, Otsuka, Abbott. P.M. has received a research grant from Vifor Pharma and Fonds Wetenschappelijk Onderzoek (grant number: 1127917 N) and consultancy fees from AstraZeneca, Boehringer Ingelheim, Novartis and Vifor Pharma. C.M. has received personal fees and non-financial support from Roche Diagnostics; personal fees from Novartis, Cardiorentis, Boehringer Ingelheim; grants and non-financial support from Abbott; grants, personal fees and non-financial support from Singulex, BRAHMS, outside the submitted work. J.L. has received consulting and/or lecture fees from Bayer, Boehringer Ingelheim, Novartis, Pfizer, Orion Pharma and Vifor Pharma. W.H.W.T. has been supported by grants from the NIH and has served as a consultant for MyoKardia Inc, and Sequana Medical Inc. H.S. received honorarium for advisory boards and lectures from Novartis, Servier, Vifor Pharma and AstraZeneca. F.H.V. has received consultancy fees from Boehringer Ingelheim and Novartis. U.D. has received research grants from Vifor Pharma and consultancy fees from AstraZeneca, Merck and Pfizer. P.R. reports consulting for Idorsia, G3P; honoraria from AstraZeneca, Bayer, CVRx, Fresenius, Grunenthal, Novartis, NovoNordisk, Relypsa, Servier, Stealth Peptides, and Vifor Fresenius Medical Care Renal Pharma; and travel grants from AstraZeneca, Bayer, CVRx, Novartis, and Vifor Fresenius Medical Care Renal Pharma; Cofounder: CardioRenal. M.M. has received consulting honoraria from Bayer, Novartis, and Servier, and speaker's fees from Abbott Vascular and Novartis. A.M. received speaker's honoraria from Abbott, Orion, Roche and Servier; fee as member of advisory board and/or Steering Committee and/or research grant from BMS, Adrenomed, Neurotronik, Roche, Sanofi and Sphyngotec. P.S. has received grants/research supports from Ministry of education, science and technological development of Republic of Serbia; honoraria or consultation fees from Servier, Boehringer Ingelheim, Hemofarm, Novartis, AstraZeneca; participation in a company sponsored speaker's bureau: Fondazione Internationale Menarini. F.R. reports grants and personal fees from SJM/Abbott, Novartis, Bayer, Servier; personal fees from Zoll, AstraZeneca, Sanofi, Amgen, BMS, Pfizer, Fresenius, Vifor, Roche, Cardiorentis, Boehringer Ingelheim; other from Heartware; grants from Mars, during the conduct of the study; since 1 January 2018: no personal payments/all payments directly to the University of Zurich. A.C. in the last 5 years received honoraria and/or lecture fess from Actimed, AstraZeneca, Faraday, Gore, Impulse Dynamics, Menarini, Novartis, Nutricia, Resmed, Respicardia, Servier, Stealth Peptides, Verona, Vifor. The other authors report no conflicts of interest.

