

CD163 deficiency facilitates lipopolysaccharide-induced inflammatory responses and endotoxin shock in mice
Abstract
Objectives
Septic (or endotoxin) shock is a severe systemic inflammatory disease caused by bacteraemia or endotoxaemia. Although it is known that increased serum levels of CD163 are observed in septic/endotoxin shock patients, the exact function and significance of CD163 in macrophage activation remain unclear. Therefore, in the current study, we tested whether CD163 contributes to the pathogenesis of endotoxin shock in mice.
Methods and results
In samples obtained from autopsy, the number of CD163-positive macrophages was increased in the kidney, liver, heart, bone marrow and spleen of patients who had died from septic/endotoxin shock when compared to patients who had died from other causes. The animal study revealed a significantly lower survival rate in CD163-deficient mice after lipopolysaccharide (LPS) injection. Several cytokines and oxidative stress-related molecules were significantly elevated in the sera of LPS-induced endotoxin shock mice models. Higher concentrations of IL-6, TNF-α, IL-1β, nitrite (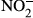 ) and nitrate (
) and nitrate (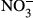 ) and a lower concentration of IL-10 were observed in CD163-deficient mice treated with LPS. Similar results were observed in CD163-deficient LPS-stimulated macrophages. Furthermore, in an antitype II collagen antibody-induced arthritis (CAIA), rheumatoid arthritis model, inflammation and bone erosion scores as well as the expression of IL-6 and IL-1β were significantly increased in CD163-deficient mice.
) and a lower concentration of IL-10 were observed in CD163-deficient mice treated with LPS. Similar results were observed in CD163-deficient LPS-stimulated macrophages. Furthermore, in an antitype II collagen antibody-induced arthritis (CAIA), rheumatoid arthritis model, inflammation and bone erosion scores as well as the expression of IL-6 and IL-1β were significantly increased in CD163-deficient mice.
Conclusions
CD163 was suggested to be involved in the regulation of inflammatory cytokine expression in septic/endotoxin shock and CAIA.
Introduction
Septic/endotoxin shock, a severe systemic inflammatory disease caused by bacteraemia or endotoxaemia, is associated with a high mortality rate.1, 2 Infections in the respiratory tract, genitourinary organs and intestines are common causes of septic or endotoxic shock.3 Cancer patients, especially patients with leukaemia, are at a high risk of sepsis because of immunosuppression and chemotherapy.4, 5 The inflammatory response associated with septic/endotoxin shock is predominantly caused by the cytokine storm involving activated immune cells, of which macrophages are considered to be centrally involved in the initiation of septic/endotoxin shock.6, 7 In vivo studies have demonstrated that suppression of macrophage activation improves the survival rate of septic/endotoxin shock murine models.8, 9 Therefore, investigation of the detailed mechanism of macrophage activation in septic/endotoxin shock is important for improving clinical outcomes in humans.
CD163 is a scavenger receptor specifically expressed on the cell surface of macrophages.10 CD163 binds to the haemoglobin/haptoglobin complex, bacteria and tumor necrosis factor-like weak inducer of apoptosis (TWEAK).11, 12 Several studies have reported that CD163-expressing macrophages play significant roles in malignant tumors, autoimmune diseases and lung diseases. However, few details regarding the mechanism of CD163 involvement in these diseases are known.13-15 CD163 expression is up-regulated on circulating monocytes in the peripheral blood of septic/endotoxin shock patients.16, 17 In cultured macrophages, lipopolysaccharide (LPS) exposure induces CD163 expression.18 Serum levels of macrophage markers, including CD163, are increased in patients who have died from pneumococcal bacteraemia when compared to survivors.19, 20 Many studies have focused on CD163 in the pathogenesis of septic/endotoxin shock, yet the mechanisms related to CD163 and macrophage activation have not been fully uncovered. We suggest that CD163 plays a protective role against septic/endotoxin shock, since macrophages expressing high levels of CD163 are known to have anti-inflammatory functions as M2-like macrophages.13, 14
In addition, targeted delivery of drugs used to treat septic shock via an anti-CD163 antibody has been suggested as a promising therapeutic approach in a pig model.21 The activation of CD163-expressing macrophages appears to play a significant role not only in the pathogenesis of septic/endotoxin shock but in other diseases as well. In the present study, we examined CD163 expression on macrophages in various organs obtained from the autopsy of patients who had died with or without septic shock. We then examined the function of CD163 in a mouse model of septic shock and an antitype II collagen antibody-induced arthritis (CAIA) model.
Results
Numbers of CD163-positive macrophages are increased in patients with septic shock
First, we investigated CD163 expression using samples obtained at the autopsy of patients who died with or without septic/endotoxin shock. Increased numbers of CD163-positive macrophages were observed in the liver, kidney, heart and bone marrow (Figure 1a–c). Interestingly, CD163-positive macrophages were detected in the glomerular area of tissue from patients with septic/endotoxin shock but not in tissue from patients who had not experienced septic/endotoxin shock (Figure 1a).

CD163−/− mice are sensitive to LPS exposure
Next, we investigated whether CD163 expression is induced by LPS stimulation in vitro and in vivo. CD163 expression was observed by Western blot analysis in mouse peritoneal macrophages stimulated with LPS. Weak CD163 expression was observed in unstimulated peritoneal macrophages, but the CD163 expression was significantly up-regulated in LPS-stimulated cells starting at 4 h after stimulation (Figure 2a). The CD163 expression peaked 12 h after stimulation, and its expression intensity was observed up to 48 h after stimulation (Figure 2a). LPS stimulation also increased the CD163 expression in mouse splenic macrophages, whereas weak CD163 expression was observed in unstimulated splenic control macrophages (Figure 2b). Similar results were observed in the spleens of LPS-treated mice (Figure 2c), suggesting that CD163 plays a significant role in inflammatory responses.

CD163−/− mice are sensitive to LPS exposure
Next, CD163+/+ and CD163−/− mice were then injected with LPS to investigate the function of CD163 in modulating LPS-induced inflammation. Defective CD163 expression in CD163−/− mice was confirmed by PCR, Western blot analysis and immunohistochemistry (Figure 3a–c). In the spleen, Iba-1 macrophage density was not altered by LPS treatment (Figures 2c and 3c). However, CD163 expression and CD163-positive cells were increased by LPS treatment (Figures 2c and 3c). As shown in Figure 3d, CD163-deficient mice exhibited a significantly higher mortality rate than WT control mice. Serum levels of cytokines such as TNF-α, IL-6, IL-1β and IL-10 were evaluated in CD163+/+ and CD163−/− mice following LPS injection. Compared with WT mice, CD163-deficient mice exhibited higher serum levels of TNF-α, IL-6 and IL-1β but lower levels of IL-10 (Figure 3e). The serum concentration of soluble CD163 was also increased after LPS injection in CD163+/+ mice (Figure 3f). With regard to reactive oxygen species, 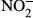 and
and 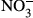 were increased in mice treated with LPS, and a significantly higher concentration was observed in CD163−/− mice (Figure 3g). SOD activity was not different between CD163+/+ and CD163−/− mice (Figure 3g). There was no difference in Hb concentration (Figure 3g).
were increased in mice treated with LPS, and a significantly higher concentration was observed in CD163−/− mice (Figure 3g). SOD activity was not different between CD163+/+ and CD163−/− mice (Figure 3g). There was no difference in Hb concentration (Figure 3g).

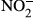 )/nitrate (
)/nitrate (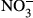 ), superoxide dismutase (SOD) and Hb at 24 h after LPS injection were measured. A one-way ANOVA test was performed for statistical analysis.
), superoxide dismutase (SOD) and Hb at 24 h after LPS injection were measured. A one-way ANOVA test was performed for statistical analysis.CD163 deficiency increased the production of inflammatory cytokines and TLR4 signalling activation, while reducing anti-inflammatory cytokine production in macrophages
To investigate the relationship between LPS-induced inflammation and CD163, cytokine expression was evaluated in LPS-stimulated peritoneal macrophages isolated from CD163+/+ and CD163−/− mice. Compared with peritoneal macrophages from CD163+/+ mice, the secretion of TNF-α, IL-6 and IL-1β was higher in peritoneal macrophages from CD163−/− mice, whereas IL-10 secretion was lower (Figure 4a). Furthermore, LPS-induced TLR4 signalling components such as p38, NF-κb and JNK were up-regulated in CD163−/− macrophages than those in CD163+/+ macrophages. (Figure 4b). These data indicate that CD163 deficiency aggravates LPS-induced inflammation and that CD163 regulates LPS-induced inflammation. The transcription of genes encoding various cytokines and other inflammation-associated molecules was evaluated using cultured macrophages, microarray analysis and quantitative real-time polymerase chain reaction (qPCR). Gene microarray analyses revealed that the expression of inflammation-related markers, including NOS2, IFN-γ, IL-2 and GBP5, was up-regulated in CD163-deficient macrophages, whereas the expression of anti-inflammatory genes, including Arg1, Ptgs1 and Mrc1, was down-regulated (Figure 4c, Supplementary table 1). The results of qPCR analyses were similar to the results of cytokine secretion assays (Figure 5). In addition, CD163-deficient macrophages stimulated with LPS exhibited up-regulated expression of CCR2 and COX2 and down-regulated expression of CCL2 (Figure 5).


CD163 deficiency exacerbates disease severity in autoantibody-/LPS-induced arthritis
Our data indicate that CD163-related signalling modulates LPS-induced macrophage activation. Thus, we also examined the role of CD163 in LPS-induced rheumatoid arthritis (RA) using a mouse model. CD163 expression was restricted to Iba-1-positive synovial tissue-resident macrophages in arthritic ankle joint tissues (Figure 6a). Double immunostaining of CD163 and Iba-1 indicated that a part of Iba-1-positive macrophages expressed CD163 (Figure 6b). To explore the role of CD163-positive macrophages in an experimental animal arthritis model generated by using antitype II collagen antibody and LPS treatment, we examined CAIA mice. CAIA is T and B lymphocyte-independent model of the effector phase of arthritis. Joint inflammation is enhanced by the inclusion of LPS treatment into the CAIA protocol.22 Clinical scores were significantly worse in arthritic CD163−/− mice when compared to their CD163+/+ counterparts (Figure 6c). Histomorphometric quantification of arthritic changes in the joint tissues confirmed the clinical assessment, with significant increases in inflammation and higher bone erosion scores in CD163−/− mice (Figure 6d). Correspondingly, IL-1β and IL-6 mRNA expression was significantly up-regulated in inflamed ankle joints obtained from CD163−/− mice when compared to joints obtained from CD163+/+ mice (Figure 6e).

Discussion
It is generally accepted that CD163 expression is up-regulated during the resolution phase of both the acute inflammatory response and chronic inflammatory disease, possibly reflecting counter-regulatory, adaptive or homeostatic mechanisms. In the present study, we demonstrated that the numbers of CD163-positive macrophages are significantly increased in several organs such as the liver, kidney, bone marrow, heart and spleen in patients with septic shock (Figure 1), and a significant role of CD163 in the pathogenesis of septic shock was suggested. Age was somewhat different between the control group (median, 66; mean, 64.8) and the septic group (median, 75; mean, 71.7). Since CD163 expression was not significantly different between ages in previous cancer patient cohort studies,22, 23 age might not affect the overexpression of CD163 in septic patients in the present study. However, as we could not use the samples from healthy patients in the present study, it is necessary to confirm CD163-overexpression in septic patients by other methods in the future. On the one hand, similar results were observed in mice with septic shock (Figure 2c and Supplementary figure 1). On the other hand, CD163-positive macrophages were not detected in the glomeruli of kidneys in any mice, regardless of LPS treatment (data not shown). CD163-positive macrophages hardly exist in the glomeruli of mouse kidneys, unlike human kidneys, under normal conditions, thus indicating that the CD163 expression in kidney macrophages differs between these two species.
Several groups have reported that patients with septic shock exhibit increased serum levels of soluble CD163. However, we believe the present study is the first to report increased numbers of CD163-positive macrophages in organs of patients with septic shock. This increase could be associated with the systemic inflammation observed in sepsis. LPS stimulation induces CD163 overexpression and neutralisation of IL-6 and/or IL-10 blocks the up-regulation of CD163 expression in human macrophages,18, 24 suggesting that CD163 overexpression is because of overproduction of IL-6 and/or IL-10 following LPS stimulation. Endotoxin is also known to induce MCP-1 production in macrophages. MCP-1 produced by resident macrophages can potentially stimulate chemotaxis of monocytes into organs, with circulating endotoxin then inducing increased expression of CD163 by infiltrating macrophages.
Although strong CD163 expression was seen in almost all Iba-1-positive macrophages in human samples (Supplementary figure S2), two populations (CD163high and CD163low/negative) were observed in LPS-stimulated macrophages by flow cytometry. This discrepancy between human and mouse macrophages might be because of the differences in gene expression between species; CD163 expression was seen in both resident macrophages and monocyte-derived macrophages in human, whereas CD163 expression was high in resident macrophages but low or negative in bone marrow-derived macrophages in mice.22, 25, 26 Genetic modifications such as methylation of the promoter region might have also affected this discrepancy, but further studies will be needed to clarify this phenomenon.
As described in the introduction, increased soluble CD163 was seen in patients who had died from bacteraemia. Increased soluble CD163 in the blood of patients with endotoxin shock was because of the shedding of the CD163 extracellular domain from monocytes/macrophages,27 and ADAM17 was found to be involved in CD163 shedding.28 A recently published study reported that CD163 expression on circulating monocytes was reduced in patients with septic shock.29 Based on these observations, we suggested that reduced CD163 expression on monocyte/macrophages through shedding was associated with the overproduction of inflammatory cytokines by macrophages, contributing to a worse clinical course in patients with septic shock.
In the present study, we demonstrated that CD163-deficient endotoxin shock mouse models exhibit a higher mortality rate associated with TNF-α, IL-1β and IL-6 overproduction and reduced IL-10 production both in vivo and in vitro (Figures 3-5). CD163, a member of the cysteine-rich scavenger receptor superfamily type B, has been identified as a haemoglobin scavenger receptor (CD163/HbSR) for the haptoglobin (Hp)-haemoglobin (Hb) complex.30 CD163 expression on circulating monocytes was shown to be significantly increased in patients who underwent cardiopulmonary bypass surgery. Further, CD163 binding the Hp-Hb complex induces IL-10 production and haem oxygenase-1 expression in monocytes, indicating that CD163 is associated with the suppression of inflammatory responses related to Hb scavenging and metabolism.31 A previous study suggested that casein kinase signalling is activated by CD163-related signalling.32 However, our unpublished experimental data do not support that conclusion. CD163 functions as a decoy receptor for TWEAK, a proinflammatory cytokine of the TNF family.33 Although we did not examine the expression of TWEAK in the present study, it is possible that TWEAK-related proinflammatory responses are linked to hypercytokinaemia in CD163−/− mice. We tried to examine the effect of LPS on casein kinase signalling in both CD163+/+ macrophages and CD163−/− macrophages in order to assess CD163-related signalling molecules. However, no difference between macrophage groups was observed (unpublished data). Thus, it remains unclear whether CD163 directly influences cytokine production by macrophages. Hb uptake via CD163 was reportedly associated with haem oxygenase-1 (HO-1) in macrophages,34 yet there was no direct link between HO-1 and cytokine production, including that of IL-6. Since the expression of some genes, such as CCR2 and COX2, was different between CD163+/+ and CD163−/− macrophages, unknown molecules, other than CD163, might regulate cytokine production. The detailed mechanism remains to be elucidated, and further studies are required to clarify the role of CD163-related signalling in macrophages. Soluble CD163, the extracellular domain that is shed from monocytes/macrophages, is known to be increased in the blood of patients with endotoxin shock.27 The function of soluble CD163 was not tested in the present study. Further studies will be needed to clarify the significance of soluble CD163 in endotoxin shock.
RA, a chronic autoimmune disease primarily affecting the joints, is characterised by chronic proliferative synovitis. Infiltration of macrophages, fibroblasts, lymphocytes and dendritic cells occurs in synovial tissues, and the degree of macrophage infiltration in RA is highly correlated with articular destruction.35, 36 Therapies involving anti-IL-6 and anti-TNF-α antibodies improve the clinical outcome of RA,37 and these cytokines are secreted by macrophages. RA patients exhibit increased levels of CD163 in serum, which correlate with disease activity.38 Increased CD163 expression was observed in synovial tissues of RA patients with significant inflammation.39 In the present study, CD163 deficiency was associated with exacerbated clinical symptoms of the RA mouse model (Figure 6), suggesting a protective role for CD163-related signalling in the pathogenesis of RA. In the present CAIA model, LPS was used to activate TLR4 signalling which also plays a role in human RA.40 An increased RA clinical score in cases of CD163 deficiency might be because of the acceleration of TLR4 signalling in arthritis.
In conclusion, the results of the present study suggest that CD163-related signalling plays a protective role through suppressing inflammatory responses in septic shock patients. Although more detailed studies are required to fully elucidate the role of CD163 in RA, the current results indicate that this molecule plays a protective role. Stimulation of CD163 might be a promising approach for treating both septic/endotoxin shock and RA.
Methods
Tissue samples
We evaluated paraffin-embedded tissue samples from nine autopsy cases of patients who had died from septic/endotoxin and seven control autopsy cases of patients who had died without septic/endotoxin shock. Autopsies were performed at Kumamoto University Hospital between 2012 and 2017 (Supplementary table S2). The Institutional Review Board at Kumamoto University approved this retrospective study (approval no. 2224). Immunohistochemical analysis of CD163 and induction of brown adipocytes (Iba-1, a pan-macrophage marker) was performed using freshly prepared 3-µm sections as described in a previous study.22 Briefly, sections underwent heat-induced antigen retrieval and were then treated with primary antibodies against CD163 (×200 dilution, NCL-L-CD163; Leica Biosystems, Nussloch, Germany), followed by HRP-labelled anti-mouse immunoglobulin (×1 dilution, code424132; Nichirei, Tokyo, Japan). Reactions were visualised with 3,3′-diaminobenzidine tetrahydrochloride (DAB, code425011 Nichirei). CD163-positive cells were counted with a microscope in four randomly selected high-power fields by two pathologists who were blinded to patient information and background data. As it was difficult to determine the cell density in the spleen because of the large number of positive cells, areas of CD163-positive staining were evaluated using ImageJ software. For double immunostaining, anti-PCNA antibody (x500 dilution, clone PC10, M0879; DAKO, Glostrup, Denmark) was used, and positive signal was visualised with the HistoGreen substrate (#AYS-E109; Linaris, Dossenheim, Germany) as the second step. For mouse tissue samples, an anti-CD163 antibody (clone EPR19518, ×1000 dilution, ab182422; Abcam, Cambridge, UK) and an anti-Iba-1 antibody (rabbit polyclonal, ×500 dilution, 019-19741; WAKO, Tokyo, Japan) were used as primary antibodies.
Animals
CD163-deficient (CD163−/−, C57BL/6N background) mice were purchased from the Knockout Mouse Project (https://www.komp.org). Mice were backcrossed with C57BL/6N mice (CREA Japan, Shizuoka, Japan) to solve the problem of passenger mutations and SNPs observed in congenic mice,41, 42 and then CD163+/− mice were housed under specific pathogen-free conditions. Mice were genotyped as previously described.22 During the course of the experiment, we observed no significant differences in body weight between CD163−/− mice and their wild-type littermates. At 8 to 10 weeks of age, male mice were treated with LPS (Escherichia coli, L4391; Sigma, St. Louis, MO, USA) via intraperitoneal injection and monitored to assess survival for a period of up to 3 days. Survival data were plotted from pooled data of two independent procedures. All animal procedures were planned according to the ARRIVE guideline43 and approved by the Animal Research Committee at Kumamoto University (#28-003). Blood samples were collected from the orbital vein in accordance with the guideline published by the Institutional Animal Care and Use Committee and ‘ARRIVE’ guidelines (Animals in Research Reporting In Vivo Experiments). Concentration of nitrite (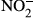 ) and nitrate (
) and nitrate (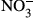 ), levels of superoxide dismutase (SOD) and Hb were measured using the NO2/NO3 assay kit (NK08; Dojindo, Kumamoto, Japan), SOD assay kit (S311; Dojindo) and Hb test kit (271-73901; Wako), respectively.
), levels of superoxide dismutase (SOD) and Hb were measured using the NO2/NO3 assay kit (NK08; Dojindo, Kumamoto, Japan), SOD assay kit (S311; Dojindo) and Hb test kit (271-73901; Wako), respectively.
Cell culture
Mouse resident peritoneal macrophages were cultured in Dulbecco's modified Eagle's medium (041-29775; WAKO) supplemented with 10% foetal bovine serum (10100; Invitrogen, Waltham, MA, USA). Dishes were rinsed with phosphate-buffered saline (PBS) to remove nonadherent cells before culture experiments.
Flow cytometry
Spleen cells were incubated in FcR blocking solution (#101319; BioLegend, San Diego, CA, USA) and then treated with an F4/80 macrophage marker FITC-labelled antibody (×200 dilution, MCA497; AbD Serotec, Oxford, UK) and anti-mouse CD163 (×500 dilution, sc-33560, rabbit polyclonal, Santa Cruz, Biotech. Dallas, TX, USA). A PE-labelled anti-rabbit antibody (×100 dilution, 406421; BioLegend) was used as the secondary antibody. Data were analysed using FACS Verse (BD Bioscience, Franklin Lakes, NJ, USA).
Western blot analysis
In some experiments, CD163 expression was determined by Western blot analysis, as described previously.22 Briefly, macrophages were solubilised with Triton X-100 (9002-93-1; WAKO) and boiled for 5 min in 2% SDS and 2-mercaptoethanol (60-24-2; WAKO). Protein samples were run on a 10% SDS–polyacrylamide gel (Bio-Lad, Hercules, CA, USA) and then transferred to a PVDF membrane (IPVH00010, Merck Millipore, Burlington, MA, USA). Next, membranes were incubated with an anti-mouse CD163 antibody (ICA-TG4-RBP1; Cosmo Bio., Tokyo, Japan) and were then reblotted with an anti-β-actin antibody (sc-47778, Santa Cruz Biotech, Santa Cruz, CA, USA) as a loading control.
TLR4 signalling was also determined by western blot analysis. Briefly, macrophages were solubilised with Triton X-100 (WAKO) and boiled for 5 min in 2% SDS and 2-mercaptoethanol (WAKO). Protein samples were run on a 10% SDS–polyacrylamide gel (Bio-Lad) and were then transferred to a PVDF membrane (Merck Millipore). The membrane was then incubated with an antiphosphorylated p38 antibody (4631S; Cell Signaling Tech, Danvers, MA. USA), anti-p38 antibody (9212S; Cell Signaling Tech), antiphosphorylated JNK antibody (9251S; Cell Signaling Tech) and anti-JNK antibody (9258S; Cell Signaling Tech). The membrane was reblotted with an anti–β-actin antibody (sc-47778; Santa Cruz Biotech) as a loading control.
Activation of NF-κB was determined by Western blot analysis, as described previously.44 Briefly, the nuclear fraction was separated on a 10% SDS-polyacrylamide gel. Membranes were exposed to an anti-NF-κB p65 subunit antibody (rel A, sc-101748; Santa Cruz). Membranes were visualised by an HRP-conjugated anti-rabbit IgG antibody with ECL Western blotting detection reagent (RPN2232; GE Healthcare Japan, Tokyo, Japan). Membranes were then reblotted with an antilamin antibody (ab58529, Abcam Japan, Tokyo, Japan) as a loading control.
Enzyme-linked immunosorbent assay
The concentrations of IL-6, TNF-α, IL-1β and IL-10 in sera and culture supernatants were determined by enzyme-linked immunosorbent assay (ELISA) using commercially available kits (88-7064-22, 88-7324-22, 88-7013-22, 88-7105-88, Invitrogen, Carlsbad, CA, USA). The concentrations of circulating soluble CD163 (sCD163) in mice were determined by ELISA using commercially available kits (DY7435; R&D Systems, Minneapolis, MN, USA).
Quantitative real-time polymerase chain reaction
Total RNA was extracted using a TAKARA RNAiso Plus (9108; Takara, Shiga, Japan) and reverse-transcribed using a TAKARA PrimeScript RT reagent kit with gDNA Eraser (RR047B; Takara). qPCR was performed using a TAKARA TB Green Premix Ex Taq II (RR820B; Takara) using an ABI PRISM 7300 sequence detector (Applied Biosystems, Foster City, CA, USA). The primers used in this study are listed in Supplementary table S3. Relative quantitation of mRNA levels was normalised to the expression of β-actin as a housekeeping gene.
RT-PCR for genotyping
All PCR amplifications were performed with a Tks Gflex DNA polymerase (R060A; Takara Bio) in a T100 Thermal Cycler (Bio-Rad Japan, Tokyo, Japan). The primer sequences were as follows: wild allele, 50-ACTTTCCTTTGGTTGTTCTGTGTTC-30, 50-AGATGCCCCTTGCTATTCCTC-30, and mutant allele, 50-ACTTTCCTTTGGTTGTTCTGTGTTC-30, 50-GTCTGTCCTAGCTTCCTCACTG-30.
Induction of collagen antibody-induced arthritis (CAIA)
Mice (n = 5 or 6 per group, 7–8 weeks old) were intravenously injected with 500 μg of a mixture of anticollagen type II (CII) monoclonal antibodies (mAbs; 53100, Arthrogen-CIA mAb; Chondrex, LLC, Seattle, WA, USA).45 Two days later, mice were intraperitoneally injected with 50 μg of LPS.45 Arthritis was graded using a 0–16 clinical scale (0–4 per paw), as previously described.46 Histologic assessment was performed on paraffin-embedded sections stained with haematoxylin and eosin, and synovial inflammation and bone erosion were graded in blinded fashion on a 0–5 scale using an established system.47
Statistics
StatMate software (Atoms, Tokyo, Japan) or Prism software (GraphPad Software, San Diego, CA, USA) were used for statistical analysis of in vitro and in vivo data. All values from in vitro studies represent results from two or three independent experiments. All data from animal studies are expressed as mean ± SD. The statistical significance of differences between groups was determined using the Mann–Whitney U-test, the one-way ANOVA test and two-way ANOVA test. The normal distribution of in vitro data was confirmed by normality tests (Shapiro–Wilk test). A P-value < 0.05 was considered indicative of statistical significance. All experimental data were representative of at least two replicates.
Acknowledgments
We thank Ms Ikuko Miyakawa and Mr Takenobu Nakagawa for their technical assistance. This work was supported by JSPS KAKENHI (grant numbers: 16H05162, 16K09247, 16K10865, 18K06991 19K07459 and 19K09555).
Author Contributions
Yukio Fujiwara: Conceptualization; Data curation; Funding acquisition; Investigation; Project administration; Writing-original draft; Writing-review & editing. Koji Ohnishi: Conceptualization; Data curation; Formal analysis; Investigation. Hasita Horlad: Data curation; Formal analysis; Investigation. Yoichi Saito: Data curation; Investigation. Daisuke Shiraishi: Data curation; Investigation. Daiki Yoshii: Formal analysis. Shinjiro Kaieda: Data curation; Writing-original draft. Tomoaki Hoshino: Supervision. Yoshihiro Komohara: Conceptualization; Funding acquisition; Investigation; Writing-original draft; Writing-review & editing.
Conflict of Interest
The authors declare no conflict of interest.
