

Effects of basic calponin on the flexural mechanics and stability of F-actin†
Monitoring Editor: Pekka Lappalainen
Abstract
The cellular actin cytoskeleton plays a central role in the ability of cells to properly sense, propagate, and respond to external stresses and other mechanical stimuli. Calponin, an actin-binding protein found both in muscle and non-muscle cells, has been implicated in actin cytoskeletal organization and regulation. In this work, we studied the mechanical and structural interaction of actin with basic calponin, a differentiation marker in smooth muscle cells, on a single filament level. We imaged fluorescently labeled thermally fluctuating actin filaments and found that at moderate calponin binding densities, actin filaments were more flexible, evident as a reduction in persistence length from 8.0 to 5.8 μm. When calponin-decorated actin filaments were subjected to shear, we observed a marked reduction of filament lengths after decoration with calponin, which we argue was due to shear-induced filament rupture rather than depolymerization. This increased shear susceptibility was exacerbated with calponin concentration. Cryo-electron microscopy results confirmed previously published negative stain electron microscopy results and suggested alterations in actin involving actin subdomain 2. A weakening of F-actin intermolecular association is discussed as the underlying cause of the observed mechanical perturbations. © 2011 Wiley Periodicals, Inc
Introduction
Actin is one of the most conserved and ubiquitous proteins in eukaryotic cells. In addition to its roles in cell motility and as a partner with myosin in muscle contraction, actin is also the major component of the cellular cytoskeleton, giving the cell its structural integrity and mechanical properties ultimately facilitating the transmission and sensing of cellular forces [Janmey, 1998]. The actin cytoskeleton is mechanically connected to the extracellular matrix through integrin complexes, allowing the cell to sense and respond to extracellular forces [Goldschmidt et al., 2001; Katsumi et al., 2004]. Proper mechanotransduction is crucial to proper function on both a cellular and whole organism level. Mechanical stresses have long been known to affect microvascular remodeling [Skalak and Price, 1996]. For example, vascular smooth muscle development and function relies on proper cellular strain sensing, the failure of which is related to pathological phenotypes such as atherosclerosis and increased aortic stiffness [Mitchell et al., 2007; Hahn and Schwartz, 2009]. Bone development is also mechanosensitive, with strain rates affecting activation of bone cells and growth of new bone [Turner et al., 1995].
The actin cytoskeleton itself is a highly dynamic structure, undergoing constant assembly and disassembly through actin polymerization and depolymerization, as well as rearrangement and remodeling of existing filaments in crosslinked and bundled structures. The behavior of the cytoskeleton is highly regulated, with several known signaling pathways [Carpenter, 2000]. Crucial to this cytoskeletal remodeling are associated actin-binding proteins (ABPs), which help facilitate mechanosensing and regulate actin filament growth, severing, nucleation, and mechanics [Pollard et al., 2000; Evangelista et al., 2003; Paavilainen et al., 2004; Kim and Hai, 2005; Mierke et al., 2008; Lee et al., 2009].
After its discovery in smooth muscle [Lehman and Kaminer, 1984; Takahashi et al., 1986], calponin was suggested as a smooth muscle regulator of actomyosin [Winder and Walsh, 1990; Shirinsky et al., 1992; Winder et al., 1998; Takahashi et al., 1988a; Takahashi et al., 1988b]. In recent years, however, studies in both smooth muscle and non-muscle cells have revealed a significant involvement of calponin in cellular mechanotransduction and cytoskeletal regulation, both as an end product and as an intermediary part of mechanosensory regulatory pathways. For a recent review of the biological role of calponin, see Rozenblum and Gimona [ 2008]. Basic calponin (h1CaP) has been shown to respond to agonist-induced smooth muscle contraction by relocating from contractile actin to cortical actin [Parker et al., 1994]. This agonist-induced change was later shown to accompany cytoskeletal remodeling [Kim et al., 2008]. Acidic calponin (h3CaP) was recently shown to undergo a similar translocation from stress fibers to the cell cortex and podosome-like structures in non-muscle cells upon cell stimulation and was suggested to be involved in a regulation pathway of extracellular signal-regulated kinase 1/2 [Appel et al., 2010]. A recent study demonstrated the importance of acidic calponin in the rearrangement and regulation of the actin cytoskeleton to facilitate cell fusion [Shibukawa et al., 2010]. A growing body of work is also unveiling a role of neutral calponin (h2CaP) as a cytoskeletal regulator, itself regulated by external mechanical stimuli [Tang et al., 2006; Hossain et al., 2003; Hossain et al., 2005]. Finally, basic calponin has also been shown to cause actin bundling in vitro, which could have structural and mechanical implications for the cellular actin cytoskeleton [Kołakowski et al., 1995; Tang et al., 1997]. While only basic calponin is associated with smooth muscle cells, all three known isoforms are structurally similar [Rozenblum and Gimona, 2008], with the major differences found in the C-terminal region. While this undoubtedly gives rise to differences in exact function and interactions with other actin-associated proteins, all three isoforms share similar actin-binding regions, suggesting a common mode of actin binding across the calponin protein family.
Given calponin's strong involvement in mechanical regulation of the actin cytoskeleton, it is natural to assume that calponin has an effect on the mechanics of single actin filaments as well. In this work, we therefore studied the in vitro effects of h1CaP on F-actin to better understand the mechanism by which calponin affects the mechanical properties and dynamic turnover of single actin filaments. We imaged thermally fluctuating rhodamine-labeled actin filaments to quantify the filament flexural rigidity and observed a reduction in persistence length of calponin-decorated F-actin at low decoration densities, from 8.0 μm to 5.8 μm. We also noticed a marked reduction of filament lengths when calponin-decorated F-actin was subjected to shear, which we argue reflects increased filament shear susceptibility induced by calponin binding. This effect was exacerbated with calponin concentration, which made flexural rigidity measurements impossible at high calponin concentrations with the methods used in this work. To better understand these mechanical changes from a structural point of view, we conducted cryo-electron microscopy 3D reconstructions of calponin-decorated F-actin to determine calponin's location on actin. The results confirmed previously published negative stain electron microscopy results, revealing calponin located near actin subdomain 2, a part of actin previously implicated in regulating F-actin flexural rigidity.
Abbreviations used: ABP, actin-binding protein; BSA, bovine serum albumin; DTT, dithiothreitol; EM, electron microscopy; h1CaP, basic calponin; h2CaP, neutral calponin; h3CaP, acidic calponin.
Materials and Methods
Actin Flexural Rigidity
 (1)
(1) (2)
(2)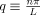 . The factor of 2 in the denominator ensures that Lp reflects the “true” persistence length of the filament, i.e., the persistence length as it would be measured for a freely flowing filament free to fluctuate in all three spatial dimensions [Greenberg et al., 2008].
. The factor of 2 in the denominator ensures that Lp reflects the “true” persistence length of the filament, i.e., the persistence length as it would be measured for a freely flowing filament free to fluctuate in all three spatial dimensions [Greenberg et al., 2008].For flexural rigidity measurements, lyophilized rabbit skeletal muscle G-actin, both unlabeled and labeled with rhodamine on random surface lysines to a labeling concentration of roughly 0.5 dyes/monomer, was purchased from Cytoskeleton (Cytoskeleton, Denver, CO) and stored at −80°C until use. Unlabeled actin was resuspended to a concentration of 5 mg/mL before experiments in G-buffer (0.2 mM CaCl2, 0.2 mM ATP, 10 mM DTT, and 5 mM Tris-HCl, pH 8.0). 20 μg rhodamine actin was resuspended in 50 μL G-buffer and incubated on ice for 1 h. Half of the resuspended rhodamine actin was polymerized at room temperature for 2 h in 50 mM KCl, 2 mM MgCl2, 1 mM ATP, and 1 mM DTT. The polymerized actin was then vortexed at low speed for 30 s with a table top vortexer to form F-actin fragments, diluted, and mixed with a fourfold excess of unpolymerized rhodamine-labeled G-actin to repolymerize filaments overnight at 4°C. The final result was 500 nM rhodamine F-actin in F-buffer (25 mM KCl, 25 mM imidazole, 1 mM EGTA, 4 mM MgCl2, 1 mM ATP, and 10 mM DTT).
Ferret h1 calponin used for flexural rigidity experiments was expressed in E. coli as described previously [Appel et al., 2010]. To generate a bacterial expression vector coding for ferret h1CaP we used the pTYB2 vector (IMPACT-CN System; New England Biolabs, Ipswich, MA) containing a C-terminal chitin binding domain. The ferret h1CaP fragment was amplified by PCR using oligonucleotides 5′-gga att cca tat gat gtc ctc tgc tca c-3′ and 5′-ctc gag ggc gga gtt ata gta gtt g-3′ and pET30b(+)-h1CaP as template. The h1CaP fragment was cloned via Ndel and Xhol restriction sites into the pTYB2 vector. The validity of the construct was confirmed by DNA sequencing. The calponin protein was then expressed in Escherichia coli BL21 cells. In brief, E. coli BL21 cell pellets were resuspended in lysis buffer (20 mM Tris-HCl, pH 8.0, 500 mM NaCl, 1 mM EDTA, and 0.1% Triton X-100) and sonicated. After centrifugation, the supernatant containing the expressed proteins was used for purification with chitin beads.
To avoid any potentially subtle artifacts from oxidative damage or other forms of protein degradation, calponin was used in flexural rigidity experiments within 2 weeks of purification. As a precaution, the amount of calponin needed for each flexural rigidity experiment was centrifuged for 30 minutes at 100,000 × g to pellet any damaged protein forming aggregates in solution. The supernatant was stored in 100 mM DTT overnight at 4°C to minimize oxidative damage. Both pellet and supernatant were inspected using a Bradford assay, showing that no calponin from a fresh (less than 2-week old) preparation pelleted during spindown (data not shown). Calponin stored in DTT was used at most 5 days after centrifugation. Labeled F-actin was similarly used at most 5 days after resuspension, after which a fresh lyophilized pellet was thawed and resuspended for use.
To make samples, microscope slides and coverslips were dipped and rinsed in 1 mM BSA twice and left to dry on the bench under a dust cover for use that same day to limit interaction between the glass and the actin. Actin and calponin were mixed in an oxygen scavenger buffer to final concentrations of 2 mM dextrose, 160 U glucose oxidase, 2 μM catalase, 10 mM DTT, 1 mM ATP, 100 mM KCl, 25 mM imidazole, 1 mM EGTA, and 4 mM MgCl2. The final concentration of rhodamine F-actin was 15 nM, while the final concentration of unlabeled G-actin was 85 nM for a total actin concentration of 100 nM, close to the barbed end critical concentration to help stabilize the filaments from depolymerization without significantly adding to their length [Isambert et al., 1995]. Calponin concentrations were kept relatively low for the purpose of flexural rigidity measurements, as calponin induced filament rupture under shear at higher concentrations, resulting in filaments too short to analyze (see Results). This was found to occur around a five times molar excess incubation of calponin in this work; flexural rigidity was therefore carried out at a four times molar excess incubation ratio. After incubating samples at room temperature for 30 min, 3 μL of the sample was deposited onto a BSA-coated microscope slide, and a BSA-coated cover slip was set on top and gently compressed to form a narrow (∼ 1–2 μm) flow chamber held together by the surface tension of the liquid. The sample was then taken immediately to the microscope for imaging. If necessary, imaging was delayed long enough for convective currents in the flow cell to settle, minimizing artifacts in filament shape due to fluid flow.
F-actin filaments freely fluctuating in two dimensions were visualized on an inverted Nikon Eclipse TE2000-U microscope with a 1.30 NA objective (Nikon, Melville, NY) using standard epifluorescence illumination. The sample temperature was kept at 30°C using a Bioptechs objective heater (Bioptechs, Butler, PA). Images of filaments 4–15 μm in length were captured using a Scion Image frame grabber (Scion, Frederick, MD) and an ICCD camera (PTI IC 310B, Birmingham, NJ). 40–80 images were captured of each filament. To avoid excessive temporal correlation between subsequent frames, images were captured 5 s apart [Gittes et al., 1993; Brangwynne et al., 2007].
The captured images were analyzed using a custom-written MatLab script (Mathworks, Natick, MA). The filaments were skeletonized by fitting a Gaussian across their width to yield a sub-pixel resolution backbone similarly to what was previously implemented [Brangwynne et al., 2007]. As filaments must be freely fluctuating in two dimensions for the analysis summarized in Eq. 2 to yield the correct persistence length, any filaments sticking to the glass surfaces during the imaging were excluded from analysis. After numerically parameterizing the filament shapes in terms of their tangent angle θ and their arc length s and decomposing this parameterization into its cosinusoidal modes, the persistence length was determined by plotting the data and fitting Eq. 2 to the range of the data following the q−2 trend (see Fig. 1). The first mode tended to show too low a variance, presumably due to some temporal correlation between frames, as has been observed in previous studies [Gittes et al., 1993]. The lower modes tended to deviate from the q−2 trend due to drift and other artifacts, to which they are vulnerable [Greenberg et al., 2008], while the higher modes were limited by the resolution noise floor [Brangwynne et al., 2007].

Persistence length analysis of a representative actin filament. Images of fluorescently labeled actin were captured 5 s apart without the actin filaments leaving the focal plane, as the filament was confined to fluctuate in two dimensions (inset). 40–80 images were captured of each filament. Once the filament was parameterized in terms of its tangent angle θ and arc length s, the parameterization was decomposed into orthogonal cosinusoidal modes, the amplitude variance of which is shown here as a function of
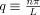 (see Materials and Methods). The persistence length was extracted by fitting Eq. 2 to the data. The higher modes were not used in fitting, as they were limited by the resolution noise floor. The lower modes were vulnerable to drift artifacts, while the lowest mode consistently exhibited low amplitude, likely due to moderate temporal correlation between frames. Scale bar is 5 μm.
(see Materials and Methods). The persistence length was extracted by fitting Eq. 2 to the data. The higher modes were not used in fitting, as they were limited by the resolution noise floor. The lower modes were vulnerable to drift artifacts, while the lowest mode consistently exhibited low amplitude, likely due to moderate temporal correlation between frames. Scale bar is 5 μm.
Actin Filament Shearing Assay
To assess the mechanical integrity of calponin-decorated actin filaments, samples with 15 nM rhodamine F-actin and 85 nM unlabeled G-actin were prepared as for flexural rigidity measurements (see above). The samples were placed on a BSA-coated microscope slide, and a BSA-coated cover slip was placed on top and compressed gently and reproducibly between samples, subjecting the actin filaments to shear as the liquid spread between the two glass slides. The resulting samples contained actin filaments whose motion was constrained to two dimensions, facilitating epi-fluorescence microscopy. Microscopy was carried out similarly to actin flexural rigidity experiments (see above). For each sample, several images were collected, and the length of each filament was measured using ImageJ. A total of 250–800 filaments were counted for each calponin concentration, ranging from no calponin to a 50 times molar excess.
SDS-PAGE Gels
To confirm calponin binding to actin, 500 nM polymerized actin was incubated with various concentrations of calponin for 30 min at room temperature in the same oxygen scavenger buffer used in flexural rigidity and actin shearing experiments and ultracentrifuged at 100,000 × g for an additional 30 min. The pellet was collected and run on a 12.5% SDS-PAGE gel. The ratios of calponin to actin were determined by staining densitometry of each gel band.
Electron Microscopy
For electron microscopy studies, skeletal muscle actin was prepared from rabbit back muscle using the method of Straub [ 1942] with the modification of Drabikowski and Gergely [ 1964]. Basic calponin used for EM studies was prepared from chicken gizzard muscle as previously described [Takahashi et al., 1986; Mezgueldi et al., 1992].
Cryo-electron microscopy samples were prepared as previously described [Xu et al., 1999]. Briefly, a few μL of filaments were applied to a carbon-coated copper grid glow-discharged in air. After 30 s the grids were blotted and immediately plunged into a 2:1 mixture of liquid ethane and propane cooled by liquid nitrogen. Samples were stored in liquid nitrogen until use.
Electron microscopy followed the methods of Xu et al. [ 1999]. Frozen grids were transferred from liquid nitrogen to a Philips CM120 cryo-electron microscope equipped with a Philips sandwich blade anticontaminator. Images of filaments over grid holes were recorded at a nominal magnification of ∼ 35,000× at 120 kV with an electron dose of 10 e−/Å2. An Eikonix 1412 CCD camera was used to digitize the micrographs. Filaments were chosen for analysis from areas close to the periphery of carbon grid holes, where ice was relatively thick and minimal evaporation presumably had occurred during the plunging of grids after blotting. Curved filaments were straightened by applying a spline-fitting algorithm [Egelman, 1986]. Helical reconstructions were carried out by standard methods [DeRosier and Moore, 1970; Amos, 1975; Amos and Klug, 1975; Owen et al., 1996] as described previously [Vibert et al., 1993, 1997]. Layer-line data extended to a resolution of about 3.5 nm as estimated by previously established methods [Owen and DeRosier, 1993]. Significant data fell within the first node of the contrast transfer function and no phase or amplitude corrections were applied [Milligan and Flicker, 1987; Milligan et al., 1990]. Maps were displayed using the program Chimera [Pettersen et al., 2004].
Results
Actin Persistence Length was Reduced When Partially Decorated with Calponin
Single actin filaments rhodamine-labeled on surface lysines were observed either in the absence of calponin or incubated with four times molar excess of h1CaP (see Materials and Methods). As our sample preparation method confined the fluctuating filaments to two-dimensional motion, each filament was imaged over several minutes using epifluorescence microscopy without the filament leaving the focal plane (Fig. 1, inset). The persistence length of each filament was extracted by fitting Eq. 2 to the data after parameterizing each filament in terms of its tangent angle θ and arc length s.
Actin labeled with rhodamine directly on surface lysines had a persistence length value of 8.0 ± 0.4 μm. We observed a relative change in persistence length when rhodamine actin was incubated with four times molar excess of h1CaP, which reduced Lp to 5.8 ± 0.2 μm, statistically significantly different from calponin-free actin (P < 0.0001 as determined by a two-tailed Student's t-test). Error bars indicate standard error of the mean, based on the number of observed filaments, which for all conditions was between 13 and 15. All measured filaments were prepared from the same lot of actin and calponin. Separate experiments on different lots of actin and preparations of calponin were also conducted. The persistence length changes were reproducible between preparations, although the binding affinity of calponin varied slightly between preparations and with calponin freshness as tested by SDS-PAGE (data not shown), and thus changed the effectiveness of calponin at lowering Lp and therefore the exact incubation ratio at which the reported changes were seen. Persistence length measurements were not conducted at higher calponin concentrations, as the resulting actin filaments were too short to undergo a persistence length analysis under these conditions (see Fig. 2 and below).

Increasing the concentration of calponin resulted in markedly shorter actin filaments after applying shear. The histograms show normalized length counts, binned into 0.5 μm groups, for calponin-free actin (100 nM actin; panel A), actin incubated with 8 times molar excess of calponin (100 nM actin and 800 nM h1CaP; panel B), and actin incubated with 50 times molar excess of calponin (100 nM actin and 5 μM h1CaP; panel C). 250–800 filaments were counted for each condition. A few actin filaments longer than 10 μm were observed in samples without calponin and were included to correctly normalize the data, although not shown in the histogram. Insets show representative fluorescence microscopy images of each condition. Scale bar is 10 μm.
Actin Filaments were Shorter After Applied Shear when Decorated with Calponin
When preparing samples of 100 nM actin incubated with varying amounts of calponin and compressing the glass slides together to apply shear to the actin, we noted that actin filaments were shorter when incubated with calponin. This effect was exacerbated with calponin concentration over the range investigated (0–5 μM h1CaP, corresponding to 0 to 50 times molar excess). The shown filament lengths were measured immediately after sample preparation. Figure 2 shows the length distributions of calponin-free actin filaments (panel A), and of filaments incubated with 0.8 and 5 μM calponin (panels B and C, respectively). The insets show representative fluorescence microscopy images of filaments under each condition. The length distributions at each concentration were not observed to evolve over time.
Amount of Calponin Bound to Actin was Estimated by SDS-PAGE
To confirm calponin binding to actin and to estimate the extent of calponin decoration on actin in the fluorescence microscopy experiments, actin was incubated with calponin at varying concentrations under identical buffer conditions to the flexural rigidity and shearing experiments (see Materials and Methods) and ultracentrifuged at 100,000 × g. The pellet was collected and run on a 12.5% SDS-PAGE gel to estimate binding ratios. Three different incubation ratios (0:1, 5:1, and 10:1 h1CaP: actin) were tested using 500 nM of rhodamine F-actin (Fig. 3, lanes 4–6). The gel bands were identified by a Biorad standard (lane 1) and two control lanes containing only actin (lane 2) or h1CaP (lane 3). The binding ratios at the different incubation ratios were determined by densitometry and are indicated as h1CaP:actin above each lane on Figure 3.

SDS-PAGE gel showed sub-saturating binding of calponin to actin. F-actin was incubated with basic calponin under conditions similar to those of the flexural rigidity and actin shearing experiments (see Materials and Methods) and ultracentrifuged at 100,000 × g, and the pellet was run on a 12.5% SDS-PAGE gel. 1: BioRad standard 161–0363, with molecular weight markers shown to the left; 2: control lane with 4 μg actin; 3: control lane with 4 μg h1CaP; 4–6: 500 nM F-actin incubated and pelleted with 0, 2.5 μM, or 5 μM h1CaP. Molar binding ratios derived from densitometry measurements are indicated as h1CaP:actin above each lane. The results show that persistence length changes occurred at sub-saturating calponin binding, while filament rupture persisted to near full decoration.
Cryo-Electron Microscopy Reconstructions Showed Calponin Density Over Actin Subdomain 2
To elucidate the structural interactions between basic calponin and actin, and to confirm previous negative stain electron microscopy results, cryo-electron microscopy images of calponin-decorated actin were used to generate a spatial reconstruction of the filament. Figure 4 shows the density map of actin (green) with calponin (pink) over actin subdomain 2, the smallest of the four actin subdomains, bridging subdomains 1 of longitudinally neighboring actin monomers. The location of calponin on actin determined by these cryo-EM reconstructions is in agreement with previous negative stain electron microscopy work on calponin-actin [Hodgkinson et al., 1997] and places calponin within range of regions important for actin-actin contacts, including the DNAse I binding loop of actin subdomain 2.

Cryo-electron microscopy of calponin-decorated actin revealed calponin density over actin subdomain 2. Shown is the difference map of actin (green) with the calponin density (pink) over subdomain 2, bridging subdomains 1 of longitudinally neighboring actin monomers. The four subdomains of one monomer, as well as subdomain 1 of the neighboring monomer, are labeled for spatial reference.
Discussion
We investigated the effect of basic calponin on actin flexural mechanics and stability. In addition to a ∼ 25% reduction in flexural rigidity at low calponin incubation ratios, calponin binding also resulted in consistently shorter filaments after filaments were subjected to shear (Fig. 2), an effect exacerbated by increasing calponin concentrations. SDS-PAGE gels of cosedimented actin and calponin suggested that the observed actin flexural rigidity changes occurred at partial calponin decoration, while the shear-induced filament shortening persisted until calponin saturation (Fig. 3).
The calponin-induced reduction in flexural rigidity could either originate from local inhomogeneous changes, i.e. local regions of the filament of increased flexibility – or “kinks” – induced at or near calponin binding sites, or as structural changes propagated throughout the filament itself. We did not observe any local regions of the filaments with larger angular variations indicating local kinks in the filament (data not shown), although such kinks would have to be separated by several hundred actin monomers to be resolvable in our experiment, assuming 370 monomers/μm [Huxley and Brown, 1967]. We do note that a cooperative unit within actin of such a scale has been previously suggested [e.g., Orlova et al., 1995].
While calponin bound to F-actin has previously been visualized by electron microscopy, it remains somewhat ambiguous exactly how and where calponin binds to actin, and whether it binds in a unique fashion or through multiple modes of binding [Galkin et al., 2006]. We therefore conducted cryo-electron microscopy in an attempt to confirm previous results obtained by negative stain electron microscopy of calponin-actin [Hodgkinson et al., 1997] and found good agreement with these previously reported results (Fig. 4). In particular, cryo-electron microscopy confirmed a calponin density over subdomain 2 of actin, a common ABP targeting site [McGough, 1998; Dominguez, 2004]. Furthermore, subdomain 2 is known to be a relatively malleable part of actin, and structural changes in this subdomain have long been proposed to be a likely mechanism by which the flexibility of F-actin can be regulated [Orlova and Egelman, 1993; Bremer et al., 1994; Isambert et al., 1995]. In this light, it is not too surprising to see calponin, with its significant density over this subdomain, affecting the flexibility of F-actin. Interestingly, calponin is in contact with longitudinally adjacent monomers by reaching from subdomain 1 across subdomain 2 of one monomer to the edge of subdomain 1 of the neighboring monomer. The connection between these subdomains is a known region of intermonomeric hydrophobic contact, as a hydrophobic region of subdomain 2 on one monomer inserts itself into a hydrophobic cleft between subdomains 1 and 3 on the longitudinally neighboring monomer [Dominguez, 2004], placing calponin in reach of important intermonomer contacts in actin. Calponin also reaches residues 43–48 in subdomain 2 [Hodgkinson et al., 1997], a region of actin previously discussed to be structurally involved in altering F-actin flexural rigidity by changing the overall structure of subdomain 2 [Isambert et al., 1995]. The changes in calponin-decorated actin flexural mechanics together with the structural data reported in this work further point to the importance of subdomain 2 as a moderator of actin flexibility.
When actin filaments were subjected to shear, we observed a significant reduction in the length of calponin-actin filaments as compared to undecorated rhodamine actin (Fig. 2). It is unlikely that this reduction in average filament length was caused by a change in the depolymerization rates or critical concentrations of actin, as we did not observe any discernable changes in filament lengths over the course of an experiment, and as the relative length differences were apparent immediately after sample preparation. The severity of the length reduction was also lessened if the flow chamber was not compressed, but simply assembled by resting the cover slip on top of the microscope slide and letting it slowly settle under its own weight, thus lessening the shear experienced by the actin (data not shown). Furthermore, calponin has previously been reported to promote actin assembly and inhibit depolymerization through electrostatic interactions at low salt concentrations [Kake et al., 1995; Tang et al., 1997]. We therefore propose that the observed difference in filament lengths was caused by a shear-induced filament breakage to which calponin-decorated filaments are more susceptible. The exponential distribution of filament lengths observed both in the absence and presence of calponin (Fig. 2), which is characteristic of a double-stranded polymer in dynamic equilibrium [Howard, 2001], also supports this hypothesis.
A correlation between decreased actin filament rigidity and decreased filament stability has been observed for one other ABP, cofilin, which has been shown to both reduce actin rigidity and sever actin [Prochniewicz et al., 2005; McCullough et al., 2008]. Two models for cofilin severing of actin have been proposed: one in which increased flexibility allows actin monomers to explore a greater conformational space, making bond rupture within decorated regions more likely [Prochniewicz et al., 2005], and one in which severing occurs at ABP decoration boundaries due to a mismatch in mechanical properties between decorated and undecorated actin regions [De La Cruz, 2009]. This second model is strongly supported by observations of cofilin-induced actin severing activity [Yeoh et al., 2002; Andrianantoandro and Pollard, 2006], as well as recent direct visualizations of single actin filament rupture in the presence of labeled cofilin [Suarez et al., 2011]. In the case of calponin, our data showed the observed actin filament breaking being exacerbated with calponin concentration (Fig. 2), and we observed no reduction in severing as the filament neared calponin saturation, as would be the case if severing occurred at decoration boundaries rather than within regions of decoration, since the number of boundaries is reduced at high levels of decoration. The mechanism of the observed calponin-induced shear severing would therefore be one in which calponin reduces the flexural rigidity of actin, likely through an interaction with actin subdomain 2, allowing a larger conformational space to be explored. This in turn makes severing in calponin-decorated regions more likely under external shear.
A growing body of evidence is pointing to calponin as a cytoskeletal regulator within the cell. Naturally, in this complex environment, it seems likely that calponin's function is at least partly related to other ABPs. (Indeed, the cryo-EM reconstructions reported here confirmed calponin's location on actin near binding sites of a number of other ABPs.) However, this study suggests that calponin weakens actin filaments and causes filament rupture under applied shear. While other ABPs, such as tropomyosin, stabilize actin and could ameliorate the rupture reported here [Ono and Ono, 2002; Greenberg et al., 2008], calponin could have a direct effect on actin turnover in regions of the cell with tropomyosin-free actin [Geiger et al., 1981; Lin et al., 1988; DesMarais et al., 2002]. Smooth muscle cells are able to contract tens of microns in the order of a minute. Actin filaments in the cortical regions of smooth muscle cells would be subject to force under these conditions, and the targeting of h1CaP to the cortex of smooth muscle cells upon agonist stimulus [Parker et al., 1994] might help facilitate remodeling of adhesion plaque connections by facilitating filament rupture as the cells undergo contraction. Our data also suggest that calponin is able to affect filament mechanics at low binding ratios. Such a mechanism could be advantageous to the cell when needing to quickly remodel its cytoskeleton without greatly changing expression levels or translocating large quantities of protein.
In summary, we showed that sub-saturating amounts of basic calponin reduced the flexural rigidity of single actin filaments by roughly 25%. We also observed a reduction in lengths of calponin-decorated filaments when subjected to shear, a result we interpret as a calponin-induced increase in the actin filament's shear susceptibility. Our data indicated that filament shear susceptibility and filament rupture was exacerbated with calponin concentration to near full decoration. This suggests a mechanism of severing in which calponin reduces the rigidity of actin, allowing a larger conformational space to be explored, which in turn leads to severing within flexible calponin-decorated regions rather than at decoration boundaries. The exact mode of calponin binding and actin rupture could be further elucidated by directly visualizing fluorescently labeled calponin as it binds to actin, similarly to recent experiments with cofilin [Suarez et al., 2011]. Measuring the torsional rigidity of calponin-decorated F-actin would also further clarify whether an increased torsional flexibility could be related to the mechanical stability of the filament, as appears to be the case for cofilin [Prochniewicz et al., 2005]. Cryo-EMs of calponin-actin presented in this work confirmed negative stain EM data [Hodgkinson et al. 1997] and suggested that the reduction in flexural rigidity could originate in actin subdomain 2, a region of actin emerging as a key to modulating actin mechanics, over which part of the calponin density rests. The calponin-induced reduction in actin filament stability observed in this study suggests that regulation of filament severing and turnover is closely linked to the mechanical properties of the filament, and supports the hypothesis that structural changes in actin subdomain 2 facilitated through ABPs play an essential role in both processes.
Acknowledgements
This work was supported by the NIH Program Project Grant HL86655 (PI: Kathleen G. Morgan). Mikkel Herholdt Jensen thanks Prof. Frederick C. MacKintosh for helpful discussions.

