

Silylative Kinetic Resolution of Acyclic Secondary Benzylic Alcohols Catalyzed by Chiral Guanidine Having Axial Chirality Containing a Methoxy Group
Comprehensive Summary
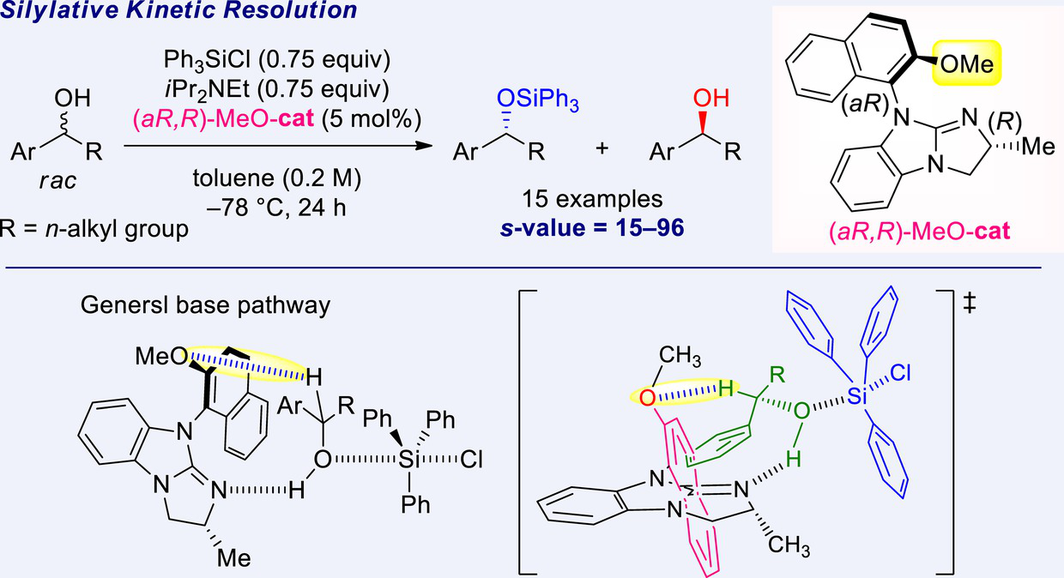
New chiral guanidine catalysts having axial chirality containing a methoxy group were synthesized. Subsequently, their catalytic ability was examined by applying the silylative kinetic resolution of racemic alcohols using chlorosilanes. Based on an X-ray crystallographic analysis of the catalysts, the functional role of the methoxy group was predicted, and the plausible reaction pathways and a transition state were described. It was also revealed that the existence of hydrogen bonding between the methoxy group on the catalyst and hydrogen atom at C-1 position of the substrates was of great importance for attaining high selectivity and reactivity. The proposed method is applicable to various acyclic aryl, heteroaryl, and normal-alkyl alcohols exhibiting medium to high s-values (s = 15–96, 15 examples).
Background and Originality Content
Silylative asymmetric protection of alcohols[1, 2] for producing chiral compounds has recently received considerable attention in organic synthesis because silyl ethers are frequently used as protection groups for hydroxy groups.[3] Since Ishikawa et al. first reported the kinetic resolution of racemic secondary alcohols via asymmetric silylation using stoichiometric amounts of chiral guanidines and silyl chlorides,[4] several characteristic catalytic silylative kinetic resolutions of racemic alcohols have been developed.[5-9] The scope of applicable substrates has been extended accordingly.
We have previously designed and synthesized chiral guanidine catalysts 1–4 to function as efficient chiral catalysts for the acylative[10] and silylative[11] kinetic resolution of racemic alcohols (Figure 1).

We have also developed the silylative kinetic resolution of a series of racemic 1-indanols using chiral guanidine catalysts 1–4 and silyl chlorides exhibiting high s-values[12] (Scheme 1).[11] The combination of catalysts and silyl chlorides is important for attaining high selectivity depending on the structure of the substituents at the C-2 position on the substrates. For example, the silylative kinetic resolution of racemic 1-indanols bearing no substituents at the C-2 position has been accomplished using a combination of either the (R,R)-2 catalyst and Ph3SiCl[11a] or the (aR,R)-4 catalyst and Ph3SiCl[11e] with high s-values. In the reaction of trans-2-alkyl-1-indanols, the combination of (R)-NMBG (1) and Ph3SiCl is the best.[11c] Furthermore, (R,R)-2 has been utilized to efficiently catalyze the kinetic resolution of 2,2-dialkyl-1-indanols using Ph2MeSiCl or PhMe2SiCl.[11b]

We have also found that when the silylative kinetic resolution of racemic acyclic benzylic alcohol rac-5, instead of cyclic benzylic alcohols, was performed under the aforementioned established conditions, only medium s-value (s = 15) was obtained (Scheme 2).[11d]

As previously mentioned, although efficient silylative kinetic resolutions of racemic alcohols have been achieved, only a few examples in which simple structures of acyclic benzylic alcohols have been applied are known (Scheme 3). Wiskur et al. reported that the silyaltive kinetic resolution of racemic acyclic benzylic alcohols using an isothiourea-type catalyst and Ph3SiCl resulted in an s-value of up to 5.6 (Scheme 3 (a)).[8a] Song et al. achieved the silyaltive kinetic resolution of racemic acyclic benzylic alcohols with s = 21–132 using low catalyst-loading of BINOL-based bis(hydroxy) polyether and HMDS (1,1,1,3,3,3-hexamethyldisilazane) (Scheme 3 (b)).[9b] Oestreich et al. performed the reaction of racemic acyclic alcohols by using Cu–H-catalyzed dehydrogenative coupling with hydrosilanes and achieved an s-value of up to 170 (Scheme 3 (c)).[5d]

In our previous studies, we synthesized axial-chirality-containing guanidine catalysts (aS,R)-3 and (aR,R)-4 by introducing a 2-methylnaphtyl group on the nitrogen atom of the guanidine groups (Scheme 4). To further determine the specific function of the catalyst, we designed novel guanidine catalysts (aR,R)-MeO-7 and (aS,R)-MeO-8 by introducing a 2-methoxynaphtyl group bearing a coordinating functional group that was expected to interact with the hydrogen atom at the C-1 position on the substrates via hydrogen bonds (Scheme 4). Herein, we report the silylative kinetic resolution of racemic acyclic benzylic alcohols catalyzed by (aR,R)-MeO-7 having axial-chirality containing a methoxy group and using chlorosilanes with high s-values.

Results and Discussion
The designed novel guanidine catalysts, (aR,R)-MeO-7 and (aS,R)-MeO-8, were synthesized based on our previous results,[10a,11e] as shown in Scheme 5. Initially, the treatment of the known benzimidazole 9[13] and (R)-alaninol in an autoclave at 130 °C for 48 h under the same reaction conditions used for preparing (R)-NMBG (1),[10a] gave the desired product 10 in low yield because of the formation of by-products. When the same reaction was carried out under milder conditions (at 80 °C for 48 h), 9 and 10 were obtained at a ratio of 82 : 18, as confirmed by 1H NMR analysis, preventing the production of unidentified by-products. The desired product 10 was obtained relatively efficiently when synthesized under the conditions of 110 °C for 120 h, which was subsequently mesylated and annulated to obtain a mixture of 7 and 8 in 38% yield and 56 : 44 ratio. Both 7 and 8 were readily separated by employing preparative thin-layer chromatography, and (aR,R)-MeO-7 in 19% yield and (aS,R)-MeO-8 in 16% yield were obtained.

To evaluate the catalytic abilities of the synthesized (aR,R)-MeO-7 and (aS,R)-MeO-8, we first applied them to the previously established silylative kinetic resolution of racemic indanol rac-11[11a,e] and compared the results with those obtained by using catalysts 1–4 (Table 1). During the silylative kinetic resolution of rac-11 with Ph3SiCl in toluene at –78 °C using 0.5 mol% of 7 and 8, both reactions proceeded smoothly to 55% conversions and exhibited high s-values (s = 53 and 33; entries 5 and 6, respectively). The highest s-value was obtained when 7 was used compared with previous results[11a,e] (entry 5 versus entries 1–4).

|
To further assess the catalytic abilities, we examined the silylative kinetic resolution of acyclic benzylic alcohol rac-5 as the target reaction (Table 2). As previously reported, the reaction using (R)-NMBG (1) yielded a medium s-value (s = 15; entry 1).[11d] No reaction occurred with (R,R)-2 (entry 2). However, when the same reaction was performed using (aS,R)-3 and (aR,R)-4 having axial chirality, high s-values were obtained (s = 91 and 70; entries 3 and 4, respectively); however, the reaction conversion was low (15% conversion) when 3 was used (entry 4). Similar examinations were conducted using novel guanidine catalysts (aR,R)-MeO-7 and (aS,R)-MeO-8 (entries 5 and 9, respectively); although both reactions proceeded smoothly to 53% and 47% conversions, respectively, medium s-value was obtained with 8 (s = 21; entry 9) and the highest s-value was obtained with 7 (s = 96; entry 5). When the reaction scale was increased from 0.2 to 1.0 mmol, almost the same results were achieved (entry 6 versus 5). The catalyst loading was decreased from 5 to 2 and 0.5 mol% (entries 5, 7 and 8). Using 2 mol% of the catalyst, the reaction proceeded with 50% conversion with an s-value of 83, which was almost the same as that obtained using 5 mol% (entry 7). Whereas on using 0.5 mol% catalyst, the conversion decreased to 40% with an s-value of 81 (entry 8).

|
To explore the scope and limitations of the present reaction, various acyclic benzylic alcohols rac-13 were applied under the optimized reaction conditions (Scheme 6). First, the reactions of phenyl alkyl alcohols 13a–13c bearing different alkyl groups were examined. Although the reactions of 13a and 13b bearing linear alkyl groups proceeded smoothly and yielded high s-values (s = 41 and 71) regardless of the alkyl chain length, no reaction occurred in the case of 13c substituted with branched alkyl group, probably due to the steric repulsion of the isopropyl group of the substrate. To explore the effect of the substituent positions, the reactions of 1-arylethanols 13d–13f containing methyl groups on the aromatic rings of the substrates were examined. The reaction of 13d with an ortho-substituent led to low conversion (17% conversion) with medium s-values (s = 15), probably due to the steric repulsion. On the other hand, the reactions of 13e and 13f with meta- and para- substituents resulted in 57% and 50% conversions with high s-values (s = 34 and 43), respectively. When similar examinations were conducted for 1-arylpropanols 13g–13i with ethyl groups instead of methyl groups, high s-values were obtained in all reactions (s = 35, 82, and 77); however, only the reaction of 13g resulted in moderate conversion (39% conversion). To further investigate the electronic nature of the substituents on the aromatic rings of the substrates, the reactions of 13j–13l were examined. Although the reaction of 13j with an ortho-substituent showed moderate reactivity (35% conversion), good selectivity was achieved in all cases (s = 39, 75, and 72), regardless of the electronic nature of the substituents. The reactions of 13m–13o containing naphthyl, pyridyl, and thiophenyl groups proceeded smoothly and led to 53%–57% conversions with high s-values (s = 39, 45, and 27). Furthermore, the importance of the aromatic rings on the substrates was highlighted by the less-reactivity of non-benzylic alcohol 13p. This indicates that the benzylic hydroxyl group is essential for the reaction to proceed.

The absolute configurations of the two synthesized compounds, 7 and 8, were determined by recrystallizing them from CH2Cl2/petroleum ether and CH2Cl2/hexane, respectively, to obtain the corresponding single crystals, which were characterized by performing X-ray crystallographic analysis. The ORTEP diagrams obtained for compounds 7 and 8 are shown in Figures 2 (a) and (b), respectively. The detailed data for these two crystals are provided in the Supporting Information (SI). The absolute configurations of 7 and 8 were found to be (aR,R) and (aS,R), respectively. Structural analysis revealed that the asymmetric unit of (aR,R)-MeO-7 consisted of two independent molecules: (aR,R)-MeO-7A (C1–C21) and (aR,R)-MeO-7B (C22–C42) (see SI). The geometric parameters of 7A and 7B in the asymmetric unit of (aR,R)-MeO-7 were confirmed to be similar. The torsion angle of (aR,R)-MeO-7A on C9–N3–C11–C20 was 68.8(3)°, and that of (aR,R)-MeO-7B on C30–N6–C32–C41 was 74.6(5)°. By contrast, the structure of (aS,R)-MeO-8 was identified to be a single molecule. The torsion angle of (aS,R)-MeO-8 on C9–N3–C11–C20 was –68.0(7)°.

To verify the stability of the diastereomers (aR,R)-MeO-7 and (aS,R)-MeO-8, each of these was exposed to toluene-d8 for 24 h or 48 h at 25, 60, and 100 °C (Scheme 7). The results revealed that both of (aR,R)-MeO-7 and (aS,R)-MeO-8 were stable at 25 °C for 24 h. At 60 °C for 24 h, the diastereomeric purities of (aR,R)-MeO-7 and (aS,R)-MeO-8 very slightly decreased to ratios (7 : 8) of 98 : 2 and 2 : 98, respectively. On the contrary, the diastereomeric purities of (aR,R)-MeO-7 and (aS,R)-MeO-8 decreased to 53 : 47 and 44 : 56, respectively, at 100 °C for 24 h. After 100 °C for 48 h, starting from either (aR,R)-MeO-7 or (aS,R)-MeO-8 also resulted in equilibrium with a 47 : 53 ratio. These results indicate that the thermodynamic stabilities of (aR,R)-MeO-7 and (aS,R)-MeO-8 are not identical.

Plausible reaction pathways and transition states based on the above results are shown in Scheme 8. The silylation of alcohols with chlorosilanes catalyzed by 4-dimethylaminopyridine (DMAP) has been proposed to function as either a Lewis base[14] or a general base.[15] The activation energies of these two pathways have been reported to be comparable; that is, either path could take place, depending on the substrate, catalyst, and other reaction conditions.[16] Conversely, in silylation of alcohols using isothiourea catalysts, isothiourea catalysts act as Lewis bases.[17, 18] In the present reaction using the chiral guanidine (aR,R)-MeO-7, we postulated that the reaction proceeds through a general base pathway (Scheme 8 (b)), not the Lewis base pathway (Scheme 8 (a)). Because the catalysts (aR,R)-4[11e] and (aR,R)-MeO-7 have similar torsion angles, it is considered that these catalysts also have similar chiral sites. High s-values were obtained for the silylative kinetic resolution of rac-5 (Table 2, entries 4 and 5). However, using (aR,R)-MeO-7 yielded a higher conversion than using (aR,R)-4. Therefore, the reaction proceeded via a general base pathway stabilized by hydrogen bonding between the oxygen atom of the methoxy group on the catalyst and hydrogen atom at the C-1 position on the substrate. The transition state via a general base pathway is illustrated in Scheme 8 (c). The catalyst abstracts the hydrogen of the hydroxyl group on the substrate while avoiding the steric hindrance from the methyl group present on the chiral center of the catalyst. The orientation of the alcohol is dictated by the stability[17d] between the guanidine cation face and aromatic ring of the substrate, exhibiting high selectivity.

Conclusions
In conclusion, we designed and synthesized guanidine catalysts having axial chirality containing a coordinating methoxy group on the nitrogen atom of the guanidine groups. The catalysts exhibited high performance in the silylative kinetic resolution of racemic acyclic aryl, heteroaryl, and normal alkyl alcohols with chlorosilanes. Diverse substrates were used for the reaction to achieve high selectivity. Further studies are being conducted to expand the substrate scope and create a specific catalyst function.
Experimental
Procedures for the silylative kinetcic resolution of rac-5 (Table 2, entry 6)
To a solution of rac-1-phenyl-1-propanol (rac-2) (136.2 μL; 1.00 mmol) in toluene (5.0 mL) at room temperature were successively added iPr2NEt (128.2 μL; 0.75 mmol) and (aR,R)-MeO-7 (16.5 mg; 0.05 mmol). After the mixture was cooled to –78 °C, Ph3SiCl (221.1 mg; 0.75 mmol) was added to it. The entire mixture was stirred for 24 h and then quenched with saturated aqueous NaHCO3 at –78 °C and diluted with EtOAc. The organic layer was separated, and the aqueous layer was extracted with EtOAc. The combined organic layers were then dried over Na2SO4. After the mixture was filtered, the solvent was evaporated to obtain the residue, which was purified by employing preparative thin-layer chromatography on silica (toluene/EtOAc = 20/1); (S)-5 (53.4 mg, 39% yield, and 99% ee) was obtained as a colorless oil and a semi-purified silyl ether. The product was re-purified by performing preparative thin-layer chromatography on silica (hexane/ EtOAc = 100/1) to achieve (R)-6 (196.2 mg, 50% yield, and 84% ee) as a colorless oil.
Acknowledgement
This work was supported by the Fukuoka Naohiko Memorial Foundation. We thank Dr. S. Mori, Division of Material Science, Advanced Research Support Center (ADRES), Ehime University, for his technical assistance.


