

Protecting-Group-Free Total Synthesis of (–)-Pallambins A—D
Xiwu Zhang
State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, 38 Xueyuan Road, Beijing, 100191 China
‡These authors contributed equally.
Search for more papers by this authorYuan Wang
State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, 38 Xueyuan Road, Beijing, 100191 China
‡These authors contributed equally.
Search for more papers by this authorPeng Chen
State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, 38 Xueyuan Road, Beijing, 100191 China
Search for more papers by this authorXinxian Cai
State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, 38 Xueyuan Road, Beijing, 100191 China
Search for more papers by this authorCorresponding Author
Yanxing Jia
State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, 38 Xueyuan Road, Beijing, 100191 China
E-mail: [email protected]Search for more papers by this authorXiwu Zhang
State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, 38 Xueyuan Road, Beijing, 100191 China
‡These authors contributed equally.
Search for more papers by this authorYuan Wang
State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, 38 Xueyuan Road, Beijing, 100191 China
‡These authors contributed equally.
Search for more papers by this authorPeng Chen
State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, 38 Xueyuan Road, Beijing, 100191 China
Search for more papers by this authorXinxian Cai
State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, 38 Xueyuan Road, Beijing, 100191 China
Search for more papers by this authorCorresponding Author
Yanxing Jia
State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, 38 Xueyuan Road, Beijing, 100191 China
E-mail: [email protected]Search for more papers by this authorMain observation and conclusion
A full account of the total synthesis of (–)-pallambins A—D (1—6) is described. The strategy was devised by simulating their biosynthetic pathway. The left-part bicyclo[3.2.1]octane system of pallambins C and D was efficiently constructed via a palladium- catalyzed oxidative cyclization. For construction of the right-part tetrahydrofuran/γ-lactone moiety (C/D rings), initial attempts to synthesize the allylic alcohol 15 for an one-step Pd-mediated alkoxycarbonylation have failed. However, during the course of this work, an unprecedented CH3Li-mediated method for conversion of bromoisoxazoline to the corresponding β-hydroxy nitrile has been discovered. Furthermore, a stepwise protocol was designed, namely an Eschenmoser-Claisen rearrangement/Lactonization to generate the C ring, and a non-classical Wittig reaction to form the D ring. During the course of this work, a palladium-catalyzed method for dehydrobromination of bromide ketone was developed. Finally, an individual transformation of pallambins C (3) and D (4) generated pallambins A (5) and B (6) under mild UV irradiation. Thus, the first enantioselective total syntheses of (–)-pallambins A—D have been achieved in 15 or 16 steps from the known chiral cyclohexenone 8. The described synthesis avoids protecting-group manipulations by designing highly chemo- and stereoselective transformations.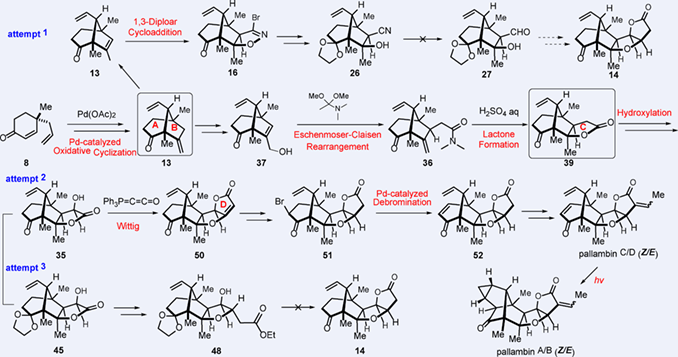
Supporting Information
| Filename | Description |
|---|---|
| cjoc202100001-sup-0001-Supinfo.pdfPDF document, 1.6 MB |
Appendix S1: Supporting Information |
Please note: The publisher is not responsible for the content or functionality of any supporting information supplied by the authors. Any queries (other than missing content) should be directed to the corresponding author for the article.
References
- 1 Wu, C.-L.; Liu, H.-J.; Uang, H.-L. A 7,8-secolabdanoid from the liverwort Pallavicinia subciliata. Phytochemistry 1994, 35, 822–824.
- 2 Toyota, M.; Saito, T.; Asakawa, Y. Novel skeletal diterpenoids from the Japanese liverwort Pallavicinia subciliata. Chem. Pharm. Bull. 1998, 46, 178–180.
- 3 Liu, H.-J.; Wu, C.-L. Neopallavicinin from the Taiwanese liverwort Pallavicinia subciliata. J. Asian Nat. Prod. Res. 1999, 1, 177–182.
- 4 Li, Z.-J.; Lou, H.-X.; Yu, W.-T.; Fan, P.-H.; Ren, D.-M.; Ma, B.; Ji, M. Structures and absolute configurations of three 7,8-secolabdane diterenes from the chinese liverwort Pallavicinia ambigua. Helv. Chim. Acta 2005, 88, 2637–2640.
- 5(a) Wang, L.-N.; Zhang, J.-Z.; Li, X.; Wang, X.-N.; Xie, C.-F.; Zhou, J.-C.; Lou, H.-X. Pallambins A and B, Unprecedented Hexacyclic 19-nor- Secolabdane Diterpenoids from the Chinese Liverwort Pallavicinia ambigua. Org. Lett. 2012, 14, 1102–1105; (b) Zhang, J.-Z.; Zhu, R.-X.; Li, G.; Wang, L.-N.; Sun, B.; Chen, W.-F.; Liu, L.; Lou, H.-X. Interconversion of the Pallambins through Photoinduced Rearrangement. Org. Lett. 2012, 14, 5624–5627.
- 6 Peng, X.-S.; Wong, H. N. C. Total synthesis of (±)-pallavicinin and (±)-neopallavicinin. Chem. Asian J. 2006, 1, 111–120.
- 7 Dong, J.-Q.; Wong, H. N. C. Biomimetic Total Synthesis of (±)- Pallavicinolide A. Angew. Chem. Int. Ed. 2009, 48, 2351–2354.
- 8 Xu, X.-S.; Li, Z.-W.; Zhang, Y.-J.; Peng, X.-S.; Wong, H. N. C. Total synthesis of (±)-pallambins C and D. Chem. Commun. 2012, 48, 8517–8519.
- 9 Ebner, C.; Carreira, E. M. Pentafulvene for the Synthesis of Complex Natural Products: Total Syntheses of (±)-Pallambins A and B. Angew. Chem. Int. Ed. 2015, 54, 11227–11230.
- 10 Huang, B.; Guo, L.; Jia, Y. Protecting-Group-Free Enantioselective Synthesis of (-)-Pallavicinin and (+)-Neopallavicinin. Angew. Chem. Int. Ed. 2015, 54, 13599–13603.
- 11 Bian, Y.-C.; Peng, X.-S.; Wong, H. N. C. Total synthesis of Pallavicinia diterpenoids: An overview. Tetrahedron Lett. 2016, 57, 5560–5569.
- 12 Martinez, L. P.; Umemiya, S.; Wengryniuk, S. E.; Baran, P. S. 11-Step Total Synthesis of Pallambins C and D. J. Am. Chem. Soc. 2016, 138, 7536–7539.
- 13 Qin, H.; Xu, Z.; Cui, Y.; Jia, Y. Total Synthesis of (±)-Decursivine and (±)-Serotobenine: A Witkop Photocyclization/Elimination/O-Michael Addition Cascade Approach. Angew. Chem. Int. Ed. 2011, 50, 4447–4449.
- 14 Li, L.; Yuan, K.; Jia, Q.; Jia, Y. Eight-Step Total Synthesis of Phalarine by Bioinspired Oxidative Coupling of Indole and Phenol. Angew. Chem. Int. Ed. 2019, 58, 6074–6078.
- 15 Liu, H.; Chen, L.; Yuan, K.; Jia, Y. A Ten-Step Total Synthesis of Speradine C. Angew. Chem. Int. Ed. 2019, 58, 6362–6365.
- 16 Zhang, X.; Cai, X.; Huang, B.; Guo, L.; Gao, Z.; Jia, Y. Enantioselective Total Syntheses of Pallambins A–D. Angew. Chem. Int. Ed. 2019, 58, 13380–13384.
- 17 Levine, S. R.; Krout, M. R.; Stoltz, B. M. Catalytic Enantioselective Approach to the Eudesmane Sesquiterpenoids: Total Synthesis of (+)- Carissone. Org. Lett. 2009, 11, 289–292.
- 18 Hong, A. Y.; Stoltz, B. M. The Construction of All-Carbon Quaternary Stereocenters by Use of Pd-Catalyzed Asymmetric Allylic Alkylation Reactions in Total Synthesis. Eur. J. Org. Chem. 2013, 2013, 2745–2759.
- 19 Ito, Y.; Aoyama, H.; Hirao, T.; Mochizuki, A.; Saegusa, T. Cyclization reactions via oxo-.pi.-allylpalladium(II) intermediates. J. Am. Chem. Soc. 1979, 101, 494–496.
- 20 Ito, Y.; Aoyama, H.; Saegusa, T. Palladium(II)-promoted cyclizations of olefinic silyl enol ethers. Preparations of .sigma.-(1-substituted 3-oxocyclopentyl)methylpalladium(II) complexes and their oxidative rearrangements. J. Am. Chem. Soc. 1980, 102, 4519–4521.
- 21 Kende, A. S.; Roth, B.; Sanfilippo, P. J. Facile, palladium(II)-mediated synthesis of bridged and spirocyclic bicycloalkenones. J. Am. Chem. Soc. 1982, 104, 1784–1785.
- 22 Kende, A. S.; Roth, B.; Sanfilippo, P. J.; Blacklock, T. Mechanism and regioisomeric control in palladium(II)-mediated cycloalkenylations. A novel total synthesis of (±)-quadrone J. J. Am. Chem. Soc. 1982, 104, 5808–5810.
- 23 Toyota, M.; Rudyanto, M.; Ihara, M. Some Aspects of Palladium-Catalyzed Cycloalkenylation: Developments of Environmentally Benign Catalytic Conditions and Demonstration of Tandem Cycloalkenylation. J. Org. Chem. 2002, 67, 3374–3386.
- 24 Toyota, M.; Wada, T.; Fukumoto, K.; Ihara, M. Total Synthesis of (±)-Methyl Atis-16-en-19-oate via Homoallyl−Homoallyl Radical Rearrangement. J. Am. Chem. Soc. 1998, 120, 4916–4925.
- 25 Toyota, M.; Odashima, T.; Wada, T.; Ihara, M. Application of Palladium-Catalyzed Cycloalkenylation Reaction to C20 Gibberellin Synthesis: Formal Syntheses of GA12, GA111, and GA112. J. Am. Chem. Soc. 2000, 122, 9036–9037.
- 26 Toyota, M.; Sasaki, M.; Ihara, M. Diastereoselective Formal Total Synthesis of the DNA Polymerase α Inhibitor, Aphidicolin, Using Palladium-Catalyzed Cycloalkenylation and Intramolecular Diels−Alder Reactions. Org. Lett. 2003, 5, 1193–1195.
- 27 Toyota, M.; Asano, T.; Ihara, M. Total Synthesis of Serofendic Acids A and B Employing Tin-Free Homoallyl−Homoallyl Radical Rearrangement. Org. Lett. 2005, 7, 3929–3932.
- 28 Nicolaou, K. C.; Tria, G. S.; Edmonds, D. J.; Kar, M. Total Syntheses of (±)-Platencin and (−)-Platencin. J. Am. Chem. Soc. 2009, 131, 15909–15917.
- 29 Varseev, G. N.; Maier, M. E. Total Syntheses of (±)-Platencin and (−)- Platencin. Angew. Chem. Int. Ed. 2009, 48, 3685–3688.
- 30 Jeker, O. F.; Carreira, E. M. Total Synthesis and Stereochemical Reassignment of (±)-Indoxamycin B. Angew. Chem. Int. Ed. 2012, 51, 3474–3477.
- 31 Yeoman, J. T. S.; Mak, V. W.; Reisman, S. E. A Unified Strategy to ent-Kauranoid Natural Products: Total Syntheses of (−)-Trichorabdal A and (−)-Longikaurin E. J. Am. Chem. Soc. 2013, 135, 11764–11767.
- 32 Sato, H.; Kusumi, T.; Imaye, K.; Kakisawa, H. Syntheses of 4-Amino-2- Hydroxybutyric Acids. Chem. Lett. 1975, 4, 965–966.
- 33 Caldirola, P.; Ciancaglione, M.; De Amici, M.; De Micheli, C. Conversion of isoxazolines to β-hydroxy esters. Synthesis of 2-deoxy-D-ribose. Tetrahedron Lett. 1986, 27, 4647–4650.
- 34 Bacher, E.; Joachim Demnitz, F. W.; Hurni, T. Studies towards the synthesis of 16α-carboxyprednisolones. Chemistry of C16-C17 fused bromoisoxazolines. Tetrahedron 1997, 53, 14317–14326.
- 35 Maimone, T. J.; Shi, J.; Ashida, S.; Baran, P. S. Total Synthesis of Vinigrol. J. Am. Chem. Soc. 2009, 131, 17066–17067.
- 36 Aouadi, K.; Jeanneau, E.; Msaddek, M.; Praly, J.-P. 1,3-Dipolar cycloaddition of a chiral nitrone to (E)-1,4-dichloro-2-butene: a new efficient synthesis of (2S,3S,4R)-4-hydroxyisoleucine. Tetrahedron Lett. 2012, 53, 2817–2821.
- 37 Ghidini, E.; Capelli, A. M.; Carnini, C.; Cenacchi, V.; Marchini, G.; Virdis, A.; Italia, A.; Facchinetti, F. Discovery of a novel isoxazoline derivative of prednisolone endowed with a robust anti-inflammatory profile and suitable for topical pulmonary administration. Steroids 2015, 95, 88–95.
- 38 Boukouvalas, J.; Fortier, G.; Radu, I.-I. Efficient Synthesis of (−)-trans- Kumausyne via Tandem Intramolecular Alkoxycarbonylation−Lactonizatio. J. Org. Chem. 1998, 63, 916–917.
- 39 Babjak, M.; Kapitán, P.; Gracza, T. The first total synthesis of goniothalesdiol. Tetrahedron Lett. 2002, 43, 6983–6985.
- 40 Hayes, P. Y.; Kitching, W. Total Synthesis and Absolute Stereochemistry of Plakortone D. J. Am. Chem. Soc. 2002, 124, 9718–9719.
- 41 Boukouvalas, J.; Pouliot, M.; Robichaud, J.; MacNeil, S.; Snieckus, V. Asymmetric Total Synthesis of (−)-Panacene and Correction of Its Relative Configuration. Org. Lett. 2006, 8, 3597–3599.
- 42 Semmelhack, M. F.; Hooley, R. J.; Kraml, C. M. Synthesis of Plakortone B and Analogs. Org. Lett. 2006, 8, 5203–5206.
- 43 Nesbitt, C. L.; McErlean, C. S. P. Total synthesis of C19 lipid diols containing a 2,5-disubstituted-3-oxygenated tetrahydrofuran. Org. Biomol. Chem. 2011, 9, 2198–2208.
- 44 Werness, J. B.; Tang, W. Stereoselective Total Synthesis of (−)-Kumausallene. Org. Lett. 2011, 13, 3664–3666.
- 45 Markovič, M.; Lopatka, P.; Koóš, P.; Gracza, T. Asymmetric Formal Synthesis of (+)-Pyrenolide D. Synthesis 2014, 46, 817–821.
- 46 Markovič, M.; Koóš, P.; Čarný, T.; Sokoliová, S.; Boháčiková, N.; Moncol’, J.; Gracza, T. Total Synthesis, Configuration Assignment, and Cytotoxic Activity Evaluation of Protulactone A. J. Nat. Prod. 2017, 80, 1631–1638.
- 47 Markovič, M.; Koóš, P.; Gracza, T. A Short Asymmetric Synthesis of Sauropunols A–D. Synthesis 2017, 49, 2939–2942.
- 48 Ikeda, R.; Kuwano, R. Asymmetric Hydrogenation of Isoxazolium Triflates with a Chiral Iridium Catalyst. Chem. - Eur. J. 2016, 22, 8610–8618.
- 49 Vyas, D. M.; Skonezny, P. M.; Jenks, T. A.; Doyle, T. W. Total synthesis of (±)-epipodophyllotoxin via a (3 + 2)-cycloaddition strategy. Tetrahedron Lett. 1986, 27, 3099–3102.
- 50 Seo, M. H.; Lee, Y. Y.; Goo, Y. M. A New Method for the Preparation of β-Hydroxy Nitriles: Transformation of 3-Bromo-2-isoxazolines to β-Hydroxy Nitriles by Treatment of Alkanethiolates. Synth. Commun. 1994, 24, 1433–1439.
- 51 Kociolek, M. G.; Kalbarczyk, K. P. Ring Opening of 3-Bromo-2-Isoxazolines to β-Hydroxy Nitriles. Synth. Commun. 2004, 34, 4387–4394.
- 52 Cheng, G.; Wang, X.; Zhu, R.; Shao, C.; Xu, J.; Hu, Y. Total Synthesis of (−)-Cocaine and (−)-Ferruginine. J. Org. Chem. 2011, 76, 2694–2700.
- 53 Aggarwal, V. K.; Angelaud, R.; Bihan, D.; Blackburn, P.; Fieldhouse, R.; Fonquerna, S. J.; Ford, G. D.; Hynd, G.; Jones, E.; Jones, R. V. H.; Jubault, P.; Palmer, M. J.; Ratcliffe, P. D.; Adams, H. Synthesis and evaluation of a broad range of chiral sulfides for asymmetric sulfur ylide epoxidation of aldehydes. J. Chem. Soc., Perkin Trans. 1 2001, 2604–2622.
- 54 Canham, S. M.; France, D. J.; Overman, L. E. Total Synthesis of (+)-Sieboldine A: Evolution of a Pinacol-Terminated Cyclization Strategy. J. Org. Chem. 2013, 78, 9–34.
- 55 Chapman, H. A.; Herbal, K.; Motherwell, W. B. Studies towards the Taming of the ‘Carbocation’ in the Regioselective Ring Opening of Epoxides to Allylic Alcohols. Synlett 2010, 2010, 595–598.
- 56 Felix, D.; Gschwend-Steen, K.; Wick, A. E.; Eschenmoser, A. Claisen'sche Umlagerungen bei Allyl-und Benzylalkoholen mit 1- Dimethylamino-1-methoxy-äthen. Helv. Chim. Acta 1969, 52, 1030–1042.
- 57
Nicolaou, K. C.; Gray, D. L. F.; Montagnon, T.; Harrison, S. T. Modulation of the Reactivity Profile of IBX by Ligand Complexation: Ambient Temperature Dehydrogenation of Aldehydes and Ketones to α,β-Unsaturated Carbonyl Compounds. Angew. Chem. Int. Ed. 2002, 41, 996–1000.
10.1002/1521-3773(20020315)41:6<996::AID-ANIE996>3.0.CO;2-I CAS PubMed Web of Science® Google Scholar
- 58 Nicolaou, K. C.; Montagnon, T.; Baran, P. S.; Zhong, Y. L. Iodine(V) Reagents in Organic Synthesis. Part 4. o-Iodoxybenzoic Acid as a Chemospecific Tool for Single Electron Transfer-Based Oxidation Processes. J. Am. Chem. Soc. 2002, 124, 2245–2258.
- 59 Nicolaou, K. C.; Zhong, Y. L.; Baran, P. S. A New Method for the One-step Synthesis of α,β-Unsaturated Carbonyl Systems from Saturated Alcohols and Carbonyl Compounds. J. Am. Chem. Soc. 2000, 122, 7596–7597.
- 60 Davis, F. A.; Vishwakarma, L. C.; Billmers, J. G.; Finn, J. Synthesis of .alpha.-hydroxycarbonyl compounds (acyloins): direct oxidation of enolates using 2-sulfonyloxaziridines. J. Org. Chem. 1984, 49, 3241–3243.
- 61 Prakash, K. R. C.; Rao, S. P. Ethoxycarbonylmethylenetriphenylphosphorane in carbohydrate chemistry, part II: a short and efficient synthesis of (+)-goniofufurone. Tetrahedron 1993, 49, 1505–1510.
- 62 Shing, T. K. M.; Tsui, H.-C.; Zhou, Z.-H. Enantiospecific Syntheses of (+)-Goniofufurone, (+)-7-epi-Goniofufurone, (+)-Goniobutenolide A, (-)-Goniobutenolide B, (+)-Goniopypyrone, (+)-Altholactone, (+)-Goniotriol, and (+)-7-Acetylgoniotriol. J. Org. Chem. 1995, 60, 3121–3130.
- 63 Kraus, G. A.; Liu, P. A Racemic Synthesis of the Novel Antibacterial Agent Juglomycin A. Synth. Commun. 1996, 26, 4501–4506.
- 64 Popsavin, V.; Grabež, S.; Popsavin, M.; Krstić, I.; Kojić, V.; Bogdanović, G.; Divjaković, V. Wittig reaction with partially protected sugar lactol derivatives. Preparation of highly cytotoxic goniofufurone analogues. Tetrahedron Lett. 2004, 45, 9409–9413.
- 65 Sun, X.-Y.; Tian, X.-Y.; Li, Z.-W.; Peng, X.-S.; Wong, H. N. C. Total Synthesis of Plakortide E and Biomimetic Synthesis of Plakortone B. Chem. - Eur. J. 2011, 17, 5874–5880.
- 66 Brennan, J.; Murphy, P. J. A phosphorane mediated synthesis of tetronic, thiotetronic and tetramic lactones. Tetrahedron Lett. 1988, 29, 2063–2066.
- 67 Schobert, R.; Dietrich, M.; Mullen, G.; Urbina-Gonzalez, J.-M. Phosphorus Ylide Based Functionalizations of Tetronic and Tetramic Acids. Synthesis 2006, 2006, 3902–3914.
- 68 Ziegler, F. E.; Tung, J. S. Synthetic studies on the macrodiolide elaiophylin. J. Org. Chem. 1991, 56, 6530–6537.
- 69 Diao, T.; Stahl, S. S. Synthesis of Cyclic Enones via Direct Palladium- Catalyzed Aerobic Dehydrogenation of Ketones. J. Am. Chem. Soc. 2011, 133, 14566–14569.
- 70 Diao, T.; Pun, D.; Stahl, S. S. Aerobic Dehydrogenation of Cyclohexanone to Cyclohexenone Catalyzed by Pd(DMSO)2(TFA)2: Evidence for Ligand-Controlled Chemoselectivity. J. Am. Chem. Soc. 2013, 135, 8205–8212.
- 71 Chen, Y.; Romaire, J. P.; Newhouse, T. R. Palladium-Catalyzed α,β-Dehydrogenation of Esters and Nitriles. J. Am. Chem. Soc. 2015, 137, 5875–5878.
- 72 Chen, Y.; Turlik, A.; Newhouse, T. R. Amide α,β-Dehydrogenation Using Allyl-Palladium Catalysis and a Hindered Monodentate Anilide. J.Am. Chem. Soc. 2016, 138, 1166–1169.
- 73 Iosub, A. V.; Stahl, S. S. Palladium-Catalyzed Aerobic Dehydrogenation of Cyclic Hydrocarbons for the Synthesis of Substituted Aromatics and Other Unsaturated Products. ACS Catal. 2016, 6, 8201–8213.
- 74 Chen, M.; Dong, G. Direct Catalytic Desaturation of Lactams Enabled by Soft Enolization. J. Am. Chem. Soc. 2017, 139, 7757–7760.
- 75 Zhao, Y.; Chen, Y.; Newhouse, T. R. Allyl-Palladium-Catalyzed α,β- Dehydrogenation of Carboxylic Acids via Enediolates. Angew. Chem. Int. Ed. 2017, 56, 13122–13125.
- 76 Huang, D.; Zhao, Y.; Newhouse, T. R. Synthesis of Cyclic Enones by Allyl-Palladium-Catalyzed α,β-Dehydrogenation. Org. Lett. 2018, 20, 684–687.
- 77 Jensen, C. M.; Chow, H.-Q.; Chen, M.; Zhai, L.; Frydenvang, K.; Liu, H.; Franzyk, H.; Christensen, S. B. Iminolactones as tools for inversion of the absolute configuration of α-amino acids and as inhibitors of cancer cell proliferation. Eur. J. Med. Chem. 2016, 114, 118–133.
- 78 Marvell, E. N.; Magoon, E. The Dienone-Phenol Rearrangement. III. Rearrangement of 6,6-Dimethyl-2,4-cyclohexadienone1. J. Am. Chem. Soc. 1955, 77, 2542–2543.
- 79 Agami, C.; Fadlallah, M.; Levisalles, J. Stéréochimie—LIV: Influence du degre de substitution de la double liaison éthylénique sur la stéréochimie de l'hydrocyanation 1,4 de cétones conjuguées. Tetrahedron 1981, 37, 909–914.
- 80 Bell, T. W. Synthesis and complexation properties of macrocyclic polyethers derived from chiral and meso-1,1'-bicyclohexyl-2,2'-diols. J. Am. Chem. Soc. 1981, 103, 1163–1171.
- 81 Sayer, J. M.; Yagi, H.; Silverton, J. V.; Friedman, S. L.; Whalen, D. L.; Jerina, D. M. Conformational effects in the hydrolyses of rigid benzylic epoxides: implications for diol epoxides of polycyclic hydrocarbons. J. Am. Chem. Soc. 1982, 104, 1972–1978.
- 82 Berkowitz, W. F.; Amarasekara, A. S.; Perumattam, J. J. Photochemical approach to the taxanes. J. Org. Chem. 1987, 52, 1119–1124.
- 83 Liu, X.; Zhang, W.; Wang, Y.; Zhang, Z.-X.; Jiao, L.; Liu, Q. Cobalt-Catalyzed Regioselective Olefin Isomerization under Kinetic Control. J. Am. Chem. Soc. 2018, 140, 6873–6882.
- 84 Hui, C.; Chen, F.; Pu, F.; Xu, J. Innovation in protecting-group-free natural product synthesis. Nat. Rev. Chem. 2019, 3, 85–107.

