

17-β estradiol attenuates the pro-oxidant activity of corticotropin-releasing hormone in macroendothelial cells
Abstract
Corticotropin-releasing hormone, which is the predominant regulator of neuroendocrine responses to stress, attenuates inflammation through stimulation of glucocorticoid release. Enhanced corticotropin-releasing hormone expression has been detected in inflammatory cells of the vascular endothelium, where it acts as a local regulator of endothelial redox homeostasis. Estrogens have beneficial effects on endothelial integrity and function, though the mechanism underlying their antioxidative effect remains as yet largely unknown. We therefore investigated the effect of 17β-estradiol on pro-oxidant action of corticotropin-releasing hormone in vitro in macroendothelial cells, and, more specifically, the role of 17β-estradiol on corticotropin-releasing hormone-induced activities/release of the antioxidant enzymes namely, endothelial nitric oxide synthase, superoxide dismutase, catalase, and glutathione. We observed that 17β-estradiol abolished the stimulatory effect of corticotropin-releasing hormone on intracellular reactive oxygen species levels and counteracted its inhibitory effect on endothelial nitric oxide synthase activity and nitric oxide release. In addition, 17β-estradiol significantly induced superoxide dismutase and catalase activity, an effect that was not significantly influenced by corticotropin-releasing hormone. Finally, 17β-estradiol significantly increased glutathione levels and the glutathione/glutathione + glutathione disulfide ratio, an action that was partially blocked by corticotropin-releasing hormone. Our results reveal that 17β-estradiol counterbalances corticotropin-releasing hormone-mediated pro-inflammatory action and thereby maintains the physiological threshold of the endothelial cell redox environment. These observations may be of importance, considering the protective role of estrogen in the development of atherosclerosis.
Abbreviations
-
- CAT
-
- catalase
-
- CRH
-
- corticotropin-releasing hormone
-
- CTL
-
- control
-
- E2
-
- 17β-estradiol
-
- eNOS
-
- endothelial nitric oxide synthase
-
- GSH
-
- glutathione
-
- GSH/GSH+GSSG
-
- glutathione/glutathione + glutathione disulfide
-
- NO
-
- nitric oxide
-
- ROS
-
- reactive oxygen species
-
- SOD
-
- superoxide dismutase
Introduction
Atherosclerosis is a chronic inflammatory disease of the arterial wall where both innate and adaptive immune responses to local stress contribute to disease initiation and progression (Geovanini and Libby, 2018). The predominant regulator of neuroendocrine responses to stress which attenuates inflammation through stimulation of glucocorticoid release, is the stress peptide corticotropin-releasing hormone (CRH) (Vale et al., 1981; Rivier and Rivier 2014; Akiba et al., 2015). In addition to the hypothalamus, CRH has been detected in inflammatory sites of peripheral tissues where its expression and concentration are enhanced in inflamed tissue fibroblasts and vascular endothelial cells (Karalis et al., 1991). Peripheral CRH acts as a local endocrine or paracrine mediator of inflammation through surface receptors, which are expressed on different tissue cells as well as the endothelium (Nazarloo et al., 2006).
An early abnormality in atherosclerosis is endothelial cell dysfunction which appears to be an indicator of vascular damage predicting both progression of atherosclerosis (Halcox et al., 2009) and incidence of cardiovascular events (Yeboah et al., 2007; Husain et al., 2015; Su, 2015). Endothelial dysfunction, a risk factor for cardiovascular disease is characterized by disturbed redox balance (Huang & Vita, 2006) and oxidative stress (Husain et al., 2015; Su, 2015). Reactive oxygen species (ROS) act as major initiators of tissue damage through up regulation of enzyme activity, signal transcription, and gene expression (Boegehold et al., 2015). However, these effects are counteracted by intracellular antioxidant defense mechanisms aimed at maintaining an optimal redox balance (Fukai and Ushio-Fukai, 2011).
Reproductive-age women have a lower incidence of cardiovascular diseases than men (Villablanca et al., 2010). This sex difference is attributed to the protective effects of estrogens on the endothelium that disappear after menopause, which is characterized by estrogen deficiency (Colditz et al., 1987; Wenger et al., 1993). Estrogens may benefit arterial integrity through different mechanisms, among which antioxidative properties (Liehr, 1996). It has been shown that in bovine aortic endothelial cells, estradiol preserves the endothelial redox balance (Cossette et al., 2013) by suppressing pro-oxidant and pro-inflammatory actions induced by a number of stress stimulators (Song et al., 2009). However, in ovariectomized spontaneous hypertensive rats, estrogen deficiency leads to increased vascular free radical production (Wassmann et al., 2001). Excessive ROS generation plays an important role in several cardiovascular diseases, including hypertension, atherosclerosis, and myocardial ischemia/reperfusion injury (Levonen et al., 2008).
In a previous study (Gougoura et al., 2010), we showed that in endothelial cells in vitro, CRH acts as a local regulator of redox balance. This action is dependent on the oxidative status of the endothelial cells. Under basal conditions, CRH increases intracellular ROS by inhibiting endothelial nitric oxide synthase (eNOS), catalase (CAT), and superoxide dismutase (SOD) activity, whereas in the presence of increased oxidative stress it acts as an anti-inflammatory factor.
CRH may act as a local regulator of endothelial redox homeostasis, while estrogens apparently have beneficial effects on endothelial integrity and function. Given that the underlying mechanism of the antioxidative effect of estrogens is still largely unknown, we investigated the effect of 17β-estradiol (E2) on the pro-oxidant action of CRH in macroendothelial cells in vitro.
Materials and methods
Reagents
CRH, 17β-estradiol (E2), fulvestrant (ICI 182.780), competitive estrogen receptor (ER) antagonist with an affinity comparable to estradiol, sodium nitrite, nitrate, nitrate reductase, nicotinamide adenine dinucleotide, hydrogen peroxide, glutathione reductase, and other reagents were obtained from Sigma-Aldrich (St Louis, MO, USA). Cell culture media Dulbecco's modified Eagle's medium (DMEM 11-965-092 and DMEM [phenol red free] 31-053-028) were obtained from Gibco (Gibco BRL, CA, USA).
Cell cultures and factors
The human endothelial cells EA.hy926 were kindly provided by Dr. Ch. Tsatsanis (University of Crete, School of Medicine, Laboratory of Clinical Chemistry, Greece). EA.hy926 cells were plated in 25 cm dishes and cultured in DMEM supplemented with 10% fetal bovine serum (FBS), 10 mM L-glutamine, 100 U/mL penicillin, and 0.1 mg/mL streptomycin (Invitrogen, CA, USA), at 37°C in a 5% CO2 atmosphere (Gougoura et al., 2010). For each experiment, confluent cells were starved in medium (phenol red free) without FBS for 24 h. Growth medium was replaced with new growth medium supplemented with CRH (10−7 M), E2 (0.5 pg/mL), and fulvestrant (5 pg/mL), a selective ERα and β antagonist (Robertson and Harrison, 2004), either individually or in combination as needed, for 2 h.
Experiments were performed with cells from passage 4. Phenol red free medium was used to minimize possible estrogenic effects of the medium, since red phenol acts as a weak estrogen (Berthois et al., 1986). All experiments were carried out in triplicate and repeated at least five times. Cell lysates were collected using a hypotonic Tris buffer, pH 7.5, in the presence of a cocktail of protease inhibitors. Protein concentrations were determined via the Bio-Rad Protein Assay (Cat.#: 5000002) according to the standard protocol using a spectrometer (Perkin Elmer UV/VIS Lambda 20; MA, USA). Culture supernatant medium was collected and both cell lysates and medium were stored at −80°C.
ROS measurement in cells
ROS levels were measured using 2,7-dichlorodihydrofluorescein diacetate (H2DCF-DA; Molecular Probes; Invitrogen Detection Technologies, CA, USA). EA.hy926 treated cells were loaded with 10 μM H2DCF-DA for 30 min at 37°C. Cells were then trypsinized, resuspended in PBS (106 cells/mL) and analyzed by fluorescence assay. Fluorescence was measured at 520 nm following excitation with 488 nm light in a VersaFluor Fluorometer (Biorad, Hercules, CA, USA).
eNOS activity assay
Catalytic activity of eNOS was determined by the conversion of L-[3H] arginine to L-[3H] citrulline. The assay was performed as previously described (Gougoura et al., 2010). Briefly, EA.hy926 cells treated with various effectors, were lysed and protein lysates (25 μg) were incubated with 1 mCi/mL L-[3 H] arginine, in the presence of 6 mM CaCl2, for 1 h at 37°C. The reaction was stopped with EDTA-HEPES buffer and scintillation fluid was added to the vials. Incorporated radioactivity was measured in a liquid scintillation b-counter (Wallace 1409). Furthermore, a standard curve of known eNOS units was carried out in order to determine the correlation between eNOS concentration and activity expressed in counts.
Measurement of nitrite (NO2−) and nitrate (NO3−) in culture supernatant media
The concentration of the NO2− and NO3− sum was determined in the culture medium. The Griess reaction was repeated after the nitrate in each specimen had been enzymatically converted to nitrite, as previously described (Gougoura et al., 2010). Aliquots of the culture media were incubated for 30 min at 37°C with nitrate reductase (10 mU) and nicotinamide adenine dinucleotide phosphohydrogenase (100 μM). Griess reagent (100 μL; 5% v/v H3PO4 with 1% w/v sulphanilic acid, and 0.1% w/v N-1-napthylethylenediamine) was added to aliquots containing 80 μL of culture medium and samples were incubated for 15 min at 37°C. Addition of the reagent converted nitrite into a deep purple azo compound, the absorbance of which was measured at 540 nm by a spectrophotometric plate reader (Biotek-Instruments Inc, Highland Park, NY, USA). The concentration of the sum of nitrite and nitrate (NO) in each sample was determined by comparing the measured absorbance to a standard curve with known sodium nitrite (NaNO2) concentrations. The lowest detection limit of the method was 0.2 μM. Each sample was analyzed in triplicates.
CAT activity determination
Catalase activity was performed as previously described (Aebi, 1984). Cell lysates (20 μg) were prepared in 67 mM phosphate buffer, pH 7.4, and incubated at 37°C for 10 min prior to measurement. Following this, 15 mM H2O2 was added and the rate of H2O2 reduction was measured in a quartz cuvette for 5 min at 240 nm. Experiments were performed in triplicates and repeated five times.
SOD activity determination
SOD catalyses the dismutation of superoxide radical (O2−) into hydrogen peroxide (H2O2), and elemental oxygen (O2). By means of Trevigen's SOD assay kit, superoxide anions convert NBT to NBT-diformazan, which absorbs light at 550 nm. SOD reduces superoxide anion concentration and thereby lowers the rate of NBT-diformazan formation. The extent of reduction in the appearance of NBT-diformazan is a measure of SOD activity present in the experimental sample. SOD activity is determined from percent inhibition of the rate (ΔΟD/min) of formation of NBT-diformazan. NBT-diformazan production was measured for 5 min. Assays were performed in triplicates and repeated five times.
Quantitative determination of glutathione (GSH) in cell lysates
The method is based on the ability of GSH to react with 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) and produce a yellow compound with extinction coefficient 13.6 mM−1 cm−1, based on Chakrapani et al. (1995). Cell lysates (10 μg) were added in 67 mM phosphate buffer pH 7.95 and DTNB 0.00396 g/mL. The optical density was measured after incubating the samples in the dark for 30 min, at 412 nm and in triplicate for each sample.
Statistical analysis
We used the GraphPad InStat Statistical package for Windows for statistical analysis. Data are expressed as mean ± standard deviation (SD). One-way analysis of variance with the Bonferroni post hoc test was used for the comparison of data and the statistical significance limit was set at P < 0.05.
Results
In order to mimic the physiological context of the oxidative stress defense system in endothelial cells, we examined the role of E2 on CRH-induced activities/release of several antioxidant enzymes, including: eNOS, NO, SOD, CAT, and GSH. The use of fulvestrant (a selective ERα and ERβ inhibitor) was to verify the participation of the ERs in any potential effects of E2 on CRH-induced activation of the antioxidant enzymes.
Effect of E2 on CRH-induced inhibition on eNOS activity and NO release
In a previous study of our group in EA.hy926 endothelial cells, we found that CRH exerts its maximum effect on NO release at the concentration of 10−7 M during 2 h incubation at 37°C (Gougoura et al., 2010). Thus, we used the same concentration in our subsequent experimental model. We also decided to use concentrations of E2 that are representative of its free physiological human levels, ranging from 0.5 to 20 pg/mL (Santanam et al., 1998). As such, EA.hy926 cells are incubated for 2 h in the presence of E2 at increasing concentrations of 0.5, 2, 5, and 10 pg/mL and eNOS activity was measured. We observed that E2 significantly increased eNOS activity and exerted its maximal effect at the lowest physiological concentration of 0.5 pg/mL (Figure 1). This effect was blocked in the presence of fulvestrant (Figure 1). Therefore, the concentration of E2 at 0.5 pg/mL was used in all our experiments.

Effect of 17β-estradio (E2) on endothelial nitric oxide synthase (eNOS) activity. After starvation for 24 h, EA.hy926 cells were incubated for 2 h at 37°C with medium alone (control cells) or medium containing E2 at 0.5 pg/mL and/or medium containing fulvestrant at 5 pg/mL. Cell lysates were collected and eNOS activity was measured. Data are shown as means ± SD of normalized data obtained in five independent experiments. All measurements were performed in triplicates. ***P < 0.001 versus control, &&&P < 0.001 versus E2.
Subsequently, the effect of 17-β estradiol on eNOS activity and NO release in the presence of CRH was investigated. We observed that CRH significantly inhibits eNOS activity and NO secretion compared with control (P < 0.001) (Figure 2). On the contrary, E2 induces a significant increase in NOS activity (P < 0.05). When endothelial cells were treated with CRH plus E2, we observed a significant increase in eNOS activity compared to control (P < 0.001) and to CRH alone (P < 0.001). This effect was abolished in the presence of fulvestrant. The changes in NO secretion were similar except that its increase in the presence of CRH plus E2 was significantly lower compared to E2 alone (P < 0.05). These results indicate that under basal conditions, E2 counteracts the inhibitory effect of CRH on endothelial NO synthase activity and 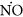 release.
release.

Effect of Ε2 and CRH on eNOS activity and NO release. After starvation, EA.hy926 cells were incubated for 2 h at 37°C in DMEM medium alone, (control cells (CTL cells)); in the presence of CRH (10−7 M) or Ε2 and/or both compounds and fulvestrant (5 pg/mL). Cell lysates were collected and eNOs activity was determined. The concentration of nitrite (NO2−) and nitrate (NO3−) in the culture media supernatant was determined. Bar graphs are presented as means ± SD of normalized data obtained in five independent experiments. All measurements were performed in triplicates. *P < 0.05 **P < 0.01 ***P < 0.001 versus CTL, +P < 0.05, ++P < 0.01 versus E2, &&&P < 0.001 versus CRH, $$P < 0.01 versus CRH + E2. CRH, corticotropin-releasing hormone; DMEM, Dulbecco's modified Eagle's medium; eNOS, endothelial nitric oxide synthase; E2, 17β-estradiol; NO, nitric oxide.
Effect of E2 on CRH-induced levels of ROS
In order to investigate the effect of 17-β estradiol on intracellular ROS content, EA.hy926 cells were incubated with E2 (0.5 pg/mL), CRH (10−7 M), and CRH + E2 simultaneously and fulvestrant (5 pg/mL). Under our culturing conditions, we observed that CRH stimulated the production of cellular ROS at significantly higher levels in comparison to control (P < 0.001) (Figure 3). On the contrary, E2 alone significantly decreased the intracellular ROS cell content compared to controls (P < 0.05) an action that disappeared when ERs were blocked by fulvestrant (Figure 3). When cells were incubated in the presence of CRH plus E2, the stimulatory effect of CRH on intracellular oxidative stress was abolished and ROS cell content reached levels which were significantly lower than those induced by E2 alone. This inhibiting effect of CRH plus E2 on intracellular ROS content was also abolished in the presence of fulvestrant.

Effect of Ε2 and CRH on intracellular ROS levels. After starvation, EA.hy926 cells, were incubated for 2 h at 37°C with medium alone (CTL cells) or medium containing E2 (0.5 pg/mL), CRH (10−7 M), and CRH + E2 simultaneously and fulvestrant (5 pg/mL). Cells were incubated with 10 μM H2DCF-DA. Relative change in mean fluorescence was calculated as the ratio of mean fluorescence for the treated to untreated (CTL) cells. The bar graph is presented as means ± SD of normalized data obtained in five independent experiments. All measurements were performed in triplicates. *P < 0.05, **P < 0.01, ***P < 0.001 versus CTL, +P < 0.05. ++P < 0.01 versus E2, &&&P < 0.001 versus CRH, $$$P < 0.001 versus CRH + E2. CRH, corticotropin-releasing hormone; CTL, control; E2, 17β-estradiol; H2DCF-DA, 2,7-dichlorodihydrofluorescein diacetate; ROS, reactive oxygen species.
Effect of E2 on CRH-mediated SOD and CAT activity
The effect of 17-β estradiol on SOD and catalase activity in the presence of CRH was also investigated. We observed that E2 alone significantly induced SOD and catalase activity compared with control (P < 0.01 and P < 0.001, respectively) and this effect was abolished in the presence of fulvestrant (Figure 4). CRH alone also significantly induced SOD and catalase activity compared with control (P < 0.001 and P < 0.05, respectively). However, the effect of E2 was more predominant on catalase activity, while CRH was predominant on SOD. The incubation of endothelial cells with CRH plus E2 was also followed by a significant increase of SOD and catalase activity compared with control (P < 0.001 and P < 0.001, respectively). The effect of E2 alone compared with CRH plus E2 on SOD activity was significantly lower (P < 0.01), while it was unchanged for CAT activity. SOD activity levels were not influenced in the presence of fulvestrant in cells treated with CRH plus E2, whereas CAT activity was significantly decreased (P < 0.01). Compared to CRH alone, the effect of CRH plus E2 on SOD activity was similar whereas on CAT activity it was significantly higher (P < 0.01). In addition, the effect of E2 or CRH on SOD1, SOD2, and catalase mRNA levels was investigated by quantitative real-time PCR; however, no statistical change was observed (Figures S1 and S2). These results may indicate that E2 does not modify induction of SOD activity by CRH but enhances catalase activity.

Effect of Ε2 and CRH on catalase activity and SOD activity. After starvation, EA.hy926 cells were incubated for 2 h at 37°C in DMEM medium alone, CTL cells; in the presence of CRH (10−7 M) or Ε2, separately or in combination, and fulvestrant (5 pg/mL). Cell lysates were prepared and analyzed for catalase activity (light blue columns) and superoxide dismutase (dark blue columns). An equal amount of proteins was used in each analysis condition. The bar graphs are presented as mean ± SD of normalized data obtained in five independent experiments. All measurements were performed in triplicates. *P < 0.05 **P < 0.01 ***P < 0.001 versus CTL, ++P < 0.01 versus E2. $$P < 0.01 versus CRH + E2. CRH, corticotropin-releasing hormone; DMEM, Dulbecco's modified Eagle's medium; E2, 17β-estradiol; SOD, superoxide dismutase.
Effect of E2 on CRH-induced changes on GSH concentration and the GSH/GSH + GSSG ratio
Finally, the effect of 17-β estradiol on GSH concentration and the GSH/GSH + GSSG ratio was investigated in the presence of CRH. We observed that EA.hy926 cells treated with CRH showed no significant changes in GSH levels, compared with untreated control cells. On the contrary, E2 provoked a large and significant increase in cellular GSH concentration (P < 0.001) and GSH/GSH + GSSH ratio (P < 0.001) that was completely abolished by the addition of fulvestrant (Figure 5). In addition, the effect of E2 or CRH on GPx mRNA levels was investigated by quantitative real-time PCR, however, no statistical change was observed (Figure S3). Co-incubation of cells with CRH plus E2, resulted in a significant increase in GSH levels and GSH/GSH + GSSH ratio compared with control and CRH treated cells (P < 0.05). However, the observed increase was far less than that induced by E2 alone (P < 0.001), indicating an inhibitory interference of CRH in the E2-induced strong stimulation of GSH levels and GSH/GSH + GSSH ratio. In addition, the complete reversal of the action of E2 alone or CRH plus E2 on GSH levels and GSH/GSH + GSSH ratio by the presence of fulvestrant indicates an estrogen-dependent effect.

Effect of CRH treatment of EA.hy926 cells on GSH levels and on reduced (GSH) and oxidized form (GSSG) of glutathione. Starved P4 EA.hy926 cells were incubated for 2 h at 37°C in DMEM medium alone, CTL cells; in the presence of CRH (10−7 M) or Ε2, separately or in combination and fulvestrant (5 pg/mL). Cell lysates were prepared and analyzed for GSH levels and the total amount of reduced (GSH) and oxidized form (GSSG) of glutathione. Bar graph is presented as means ± SD of normalized data obtained in five independent experiments. All measurements were performed in triplicates. *P < 0.05 **P < 0.01, ***P < 0.001 versus CTL, +++P < 0.001 versus E2, &P < 0.05 versus CRH, $$P < 0.01, $$$P < 0.001 versus CRH + E2. CRH, corticotropin-releasing hormone; DMEM, Dulbecco's modified Eagle's medium; CTL, control; GSH, glutathione; GSSG, glutathione disulfide.
Discussion
In the present study, we found that E2 counteracts the CRH-induced increase in intracellular free radicals and decrease in eNOS activity and NO levels, indicating an anti-inflammatory action. Moreover, E2 did not affect the CRH-induced significant SOD activity, whereas it improved the weak stimulatory effect of CRH on catalase activity. Finally, we observed that E2 had a significant stimulatory effect on GSH levels and GSH/GSH + GSSG ratio. This effect was attenuated by CRH, but remained significant compared to control conditions.
We previously showed that CRH is involved in the regulation of oxidative balance in endothelial cells in the basal state (Gougoura et al., 2010). This work suggested that CRH regulates pro-inflammatory mechanisms in endothelial cells by inducing an increase in intracellular free radicals associated with reduced eNOS activity and NO secretion, a significant increase mainly in SOD activity, and to a lesser extent in CAT activity without affecting GSH levels (Gougoura et al., 2010).
Here, we have shown that the stimulating effect of E2 on eNOS activity and NO release counteracts the inhibitory effect of CRH on endothelial NO synthase activity and NO release. This effect was abolished in the presence of the ERs antagonist, fulvestrant. Our results are consistent with findings from an earlier study in uterine artery endothelial cells, where the blockade of ERs with fulvestrant prevented estradiol eNOS activation and NO release (Chen et al., 2004). It has been suggested that E2-mediated NO production involves rapid eNOS phosphorylation and activation through MAPK and PI3K/Akt pathways (Chen et al., 1999; Chambliss et al., 2002). Rapid NO release via eNOS activation has been reported in human endothelial cells through the involvement of a plasma membrane ERα variant (ER46) which has been detected in extracts from EAhy926 endothelial cells (Russel et al., 2000; Li et al., 2003). Accumulating evidence clearly indicates the presence of membrane ERs (mERs) that transduce their effects through diverse signaling pathways mediating non-genomic events (Vivar et al., 2010). This receptor may be involved in the transduction of E2 action in our study. Interestingly, in human umbilical vein endothelial cells which express two ERα variants, the above-mentioned ERα variant transduces rapid (non-genomic) signaling whereas a 66 kDa variant transduces genomic signaling, triggered by E2 (Li et al., 2003). Signaling from mERs occurs mainly through the activation of various signal transduction pathways that are often associated with G protein-coupled receptors (GPCRs) (Lu et al., 2004), ion channels (Stefano et al., 2000), and enzyme-linked receptors.
The levels of intracellular ROS are regulated by the fine-tuning between ROS-generating factors and antioxidant enzymes, including: SOD, CAT, and glutathione peroxidase (GSH-Px). In this study, we observed that incubation of endothelial cells with E2 was followed by an increase in SOD activity and a significant increase in catalase activity. Their increase was totally abolished by inhibition of ERs by fulvestrant, indicating that E2 regulates both SOD and CAT activities through its receptors (ERα, ERβ). A similar effect of E2 on mitochondrial SOD in mice and in human aorta endothelial cells (in vitro) was previously reported (Liu et al., 2014). Endothelial cells treated with CRH significantly increased their SOD activity and to a lesser extent catalase activity. When we treated endothelial cells with CRH plus E2, we observed maximum stimulation of SOD and CAT activities. The addition of fulvestrant completely abolished only the CAT activity. This observation indicates that endothelial cells under the influence of CRH plus E2 respond differently compared to each substance alone. CRH may have the dominant effect on SOD activity, whereas regulation of CAT activity is more dependent on E2.
We also observed a simultaneous increase of SOD activity with the CRH-induced increase of intracellular free radicals, which may represent a quick cellular response in order to adjust the increased ROS levels. SOD serves as the front-line defense against ROS, implicated in the regulation of endothelial function inhibiting oxidative inactivation of NO (Fukai and Ushio-Fukai, 2011). The observed increase of endothelial SOD activity induced by E2 in this study is consistent with recently published data showing that physiological concentrations of E2 upregulate SOD gene expression (Bellanti et al., 2013). The higher stimulatory effect of CRH plus E2 on SOD activity compared to that induced by E2 alone may indicate an additive effect. However, the comparable SOD activity levels after stimulation of cells with CRH or CRH plus E2 and mainly the non-change of SOD activity after ERs blocking reduces this probability.
SOD operates in conjunction with CAT to maintain hydrogen peroxide levels resulting from SOD catalysis. In this context the increase of SOD activity induced by E2 was associated with an increase in CAT activity which was comparable to that observed with CRH plus E2. However, the observed partial inhibition of CAT activity after blockage of ERs may indicate involvement of both factors in the regulation of its enzyme activity. Considering that the catalytic reaction of CAT predominates at higher H2O2 concentrations (Kirkman and Gaetani, 2007), we can speculate that E2 may exerts its antioxidant effect at higher levels of cell oxidative stress, activating GSH.
Besides SOD and CAT, GSH is an important water-soluble antioxidant, which can directly neutralize ROS, such as lipid peroxides or H2O2, by GSH-Px consuming GSH (Coyle et al., 2006; Aquilano et al., 2014). It can be considered as “the second line defense” of the antioxidant response system, with SOD and CAT comprising the first enzymatic system that removes excessive H2O2. Here, we found that E2 increases SOD activity but increases more significantly CAT action, which confirms its ability to reduce H2O2 levels. Lower H2O2 levels are associated with a similar trend of GPx which leads to lower consumption of GSH. Thus, GSH levels are preserved and measured as an increase of GSH and ratio GSH/GSH + GSSG. This stimulatory effect of E2 on GSH levels is in accordance with that observed in H9c2 cells (Urata et al., 2006), as well as in bovine arterial endothelial cells challenged with a pro-oxidant stimulator (Dimitrova et al., 2002). In addition, it has been shown that physiological concentrations of E2 upregulate GSH-Px gene expression, mediated by MAPK (Borrás et al., 2005). Moreover, estrogen replacement therapy in premenopausal women after bilateral oophorectomy reversed the oophorectomy-induced increase of oxidative stress and the reduction in mRNA expression of GSH-Px (Bellanti et al., 2013). However, the far lesser increase in GSH levels and GSH + GSSG ratio under the action of CRH plus E2 compared to that induced by E2 may indicate an inhibitory interference of CRH on E2-induced stimulation through ERs. Probably, the presence of both E2 and CRH creates a new oxidative balance at higher levels of GSH and its ratio.
Conclusion
In conclusion, our data indicate that E2 attenuates the CRH-induced pro-oxidant effect on EAhy926 cells by stimulating eNOS activity, NO production, and the endothelial antioxidant defense system. We suggest that estrogens counterbalance the CRH-mediated pro-inflammatory action, maintaining the physiological threshold of the intracellular endothelial cell redox environment. These observations may be of importance in atherosclerosis pathogenesis considering the protective role of estrogens in the development of atherosclerosis.
Acknowledgments
The authors would like to kindly acknowledge the Faculty of Medicine of University of Thessaly who hosted the present research.
Conflict of interest
The authors declare that there are no conflict of interest.

