

Structural and thermodynamic encoding in the sequence of rat microsomal cytochrome b5†
This article concerns a protein folding problem formulated many years ago by Professor Blout. His insights into the conformational properties of peptides and proteins in their native and unfolded states and the potential for certain sequences to undergo coupled binding and refolding set the stage for modern folding studies.
Abstract
The water-soluble domain of rat microsomal cytochrome b5 is a convenient protein with which to inspect the connection between amino acid sequence and thermodynamic properties. In the absence of its single heme cofactor, cytochrome b5 contains a partially folded stretch of ˜30 residues. This region is recognized as prone to disorder by programs that analyze primary structures for such intrinsic features. The cytochrome was subjected to amino acid replacements in the folded core (I12A), in the portion that refolds only when in contact with the heme group (N57P), and in both (F35H/H39A/L46Y). Despite the difficulties associated with measuring thermodynamic quantities for the heme-bound species, it was possible to rationalize the energetic consequences of both types of replacements and test a simple equation relating apoprotein and holoprotein stability. In addition, a phenomenological relationship between the change in Tm (the temperature at the midpoint of the thermal transition) and the change in thermodynamic stability determined by chemical denaturation was observed that could be used to extend the interpretation of incomplete holoprotein stability data. Structural information was obtained by nuclear magnetic resonance spectroscopy toward an atomic-level analysis of the effects. © 2007 Wiley Periodicals, Inc. Biopolymers 89: 428–442, 2008.
This article was originally published online as an accepted preprint. The “Published Online” date corresponds to the preprint version. You can request a copy of the preprint by emailing the Biopolymers editorial office at [email protected]
INTRODUCTION
Every proteome contains a number of proteins that require a cofactor for function. In many instances, the association of this cofactor with the host polypeptide involves a large Gibbs free energy of binding and organizes the three-dimensional structure of the bound state. The conformational consequences of binding and “the adequacy of the amino acid sequence in dictating the [apoprotein] structure,” as raised by Harrison and Blout in their seminal contribution to the protein folding field,1 is the focus of intense research efforts. To this day, cofactor-induced refolding and stabilization are aspects of protein architecture that are difficult to predict on the basis of first principles. In favorable cases, in vitro experimental data can be combined with bioinformatic analyses to interpret the information coded in the primary structure. Cytochrome b5 is a convenient test protein for this purpose.
The water-soluble domain of cytochrome b5 (cyt b5) contains a single heme group used in electron transfer reactions.2-4 This group, shown in Figure 1A, is held in the protein matrix by two coordination bonds to axial histidines and by other interactions, including hydrophobic contacts and hydrogen bonds. Two forms of the holocytochrome that differ by a ˜180° rotation of the heme about its α-γ meso axis are structurally feasible. At equilibrium, these two heme orientational isomers (“A” and “B,” A representing the major form) coexist in proportions dictated by the sequence of the protein.7 The topology of holocyt b5 is mixed αβ,6 as depicted in Figure 1B, which shows the major isomer structure of the rat ferric protein.8 Because of its function and its favorable properties in solution, cyt b5 has been widely used to study the effects of amino acid replacements on the adjustment of heme chemistry.9-18

(A) The structure of the heme group (Fe-protoporphyrin IX) with Fischer nomenclature. (B) Ribbon diagram of the soluble domain of rat microsomal ferricyt b5 (NMR minimized average, 1aw3).5 The heme and the axial histidines (His39 and His63) are shown, along with the residues constituting hydrophobic core 2.6 These are Leu9 (helix 1), Trp22 (strand β4), Ile24 (strand β4), Val29 (strand β3), Ile76 (strand β2), Leu79 (strand β2), and Ile87 (helix 6). (C) The same holoprotein structure is used to mark the location of Phe35, His39, Leu46, and Tyr74. The heme group has been removed for clarity. In (B) and (C), helices are labeled H1 to H6.
The amino acid sequence of rat microsomal cyt b5 is such that the apoprotein folds into a cooperative unit of marginal stability under physiological conditions of pH and temperature.19 This unit comprises approximately two-thirds of the residues and corresponds closely to structural hydrophobic core 26; its ΔG° of unfolding is 6 kJ mol−1 at 25°C,20 which implies that the apoprotein native state is 90% populated under those conditions. The ˜30 disordered residues are located in the heme binding site; once the cofactor binds, folding proceeds further, and cyt b5 becomes a single all-encompassing cooperative unit.21 For rat microsomal ferricytochrome (ferricyt) b5, the apparent two-state ΔG° increases by 20 kJ mol−1.22
It is noteworthy that primary structure analyses23, 24 predict the 30-residue stretch to be intrinsically disordered. Thus, the sequence appears coded to prevent the formation of long-lived interactions that would require disruption to allow heme binding. At the same time, the holoprotein structure indicates that the disordered region consists of α-helical molecular recognition elements.25 A similar analysis of multiple b heme protein sequences reveals a trend for regions of low folding propensity near the heme group.26
The energetics of the cytochrome can be explored with amino acid replacements. For our purposes, these can be organized in two types. The primary structure of Type I variants contains alterations in the disordered region. These alterations, unless they cause significant refolding and extension of the structural hydrophobic core, do not affect the apparent apoprotein stability. They may, however, change that of the holoprotein, either because they lead to different direct contacts with the heme group, or because they perturb the structural propensities of elements forming the heme cavity. Type II variants contain substitutions in the folded region of the apoprotein. These can affect the stability of the protein with and without the heme group.
 ()
()In prior work,22 we used a Type I variant, containing the N57P replacement and a Glu insertion at position 59 (55TENFED→55TEPFEED, “PE” cyt b5), to inspect the relationship between holoprotein stability and heme affinity. The nuclear magnetic resonance (NMR) spectrum of ferric PE cyt b5 exhibits broad lines and indicates a marked alteration of the heme pocket structure and dynamics. The PE variant shows a loss of 6–8 kJ mol−1 in apparent holoprotein stability compared to the wild-type protein, whereas the apoprotein is unchanged. With the assumption that the heme association rate constant kH is unaffected by sequence alterations, heme transfer experiments assess the rate constant for heme loss, k−H, and therefore K−H in Eq. (1). The PE rate constant k−H is ˜25 × 10−3 min−1,22 ˜25 times faster than reported for the wild-type holoprotein.28 The acceleration corresponds to a ΔΔG° of ˜8 kJ mol−1, value within error of the apparent loss of holoprotein stability. We also observed that a Type II replacement (P81A, a location in the C-terminal helix of the holoprotein, helix 6) leads to a larger apparent effect on the apoprotein (6 kJ mol−1 decrease) than on the holoprotein (3 kJ mol−1 decrease). Heme loss from the P81A variant is intermediate between wild-type and PE variant, and too slow for accurate determination.
In this work, we continue our study of the redistribution of folding and binding energy in rat microsomal cyt b5. We destabilized the apoprotein by making a substitution directly into its cooperative core (I12A). We also explored the effects of a Type I change (N57P) to gain insight into the behavior of the PE variant and tested the limits of induced refolding with a mixed-type triple variant, F35H/H39A/L46Y. We show that it is possible to anticipate the energetic consequences of certain replacements using Eq. (1) despite unfavorable holoprotein properties.
RESULTS AND DISCUSSION
Choice of Substitutions
To destabilize the apoprotein by means of a single Type II replacement, we chose a residue in the cooperative core, but not in direct contact with the heme group. Ile12, which contributes one of the side chains of hydrophobic core 2 as shown in Figure 1B, appeared suitable. Ile12 belongs to helix 1 (H1); it is in van der Waals contact with Tyr7, His15, Trp22, Ile24, Val29, and Leu79. Inspection of the primary structures listed in the cytochrome b5 family (Pfam PF00173,29 1058 sequences at the time of this study) shows that position 12 is occupied by an isoleucine in 13% of the cases. In accordance with the requirement for a hydrophobic side chain, the other possible residues are valine (43%), leucine (33%), and phenylalanine (5%).
The PE variant was originally designed to weaken the His63 ligation bond. Two changes were combined: a glutamic acid insertion between residues 59 and 60 to offset the axial ligand and the N57P replacement to obliterate the little helical propensity present in helix 4 (H4, Thr55–Asp60).30 Heme ligation and stability are indeed affected in the PE variant,22 but the contribution of the individual changes was not assessed. To complement the existing data and probe further Type I variations, a protein containing only the N57P replacement was prepared. In wild-type sequences, an asparagine occurs at position 57 with a frequency of 2%, comparable to that of a proline. The most likely residue is an alanine (23%), and no residue type is strictly excluded. The effect of a proline at position 57 is unclear as no structure is available for the few proteins that contain it. In the cytochrome b5 domain of sulfite oxidases, a proline is located at position 56 and serves to initiate H4.31 The two proteins, however, are only 38% identical and the region in question contains a two-residue insertion rendering direct comparison difficult.
Finally, a triple variant was designed to include Type I and Type II effects. This protein harbored replacements in helix 2 (H2) (F35H and H39A, which eliminated one of the axial histidines) and in helix 3 (H3) (L46Y). The triple variant is referred to as HAY cyt b5 hereafter, and Figure 1C illustrates the location of the concerned residues. All three changes were expected to affect heme binding. The new sequence offered several possibilities; for example, heme ligation could be abolished on the side of residue 39, or a different scheme could be established with His35 or Tyr46 (or both, as observed in the hemophore HasA).32 In addition, the replacements F35H and H39A occurred in folded regions of the apoprotein, as indicated by dipolar contacts placing Phe35 in the proximity of His39, Val45 and Tyr74.19 The Pfam sequences show no instance of a histidine at position 35 and 12% occupancy of position 46 by a tyrosine. The axial His39 is present in 74% of the full set, which includes hypothetical proteins and steroid-binding domains. The expectation was a destabilized HAY apoprotein.
Effects of the Replacements on the Holoprotein—Structure
Reduced (ferrous) cyt b5 is more stable than oxidized (ferric) cyt b5.33 The latter form was chosen to bring the stability of the test proteins into an experimentally practical range. Ferricyt b5 is a S = ½ complex giving rise to well-resolved proton NMR spectra.5, 34-36 The I12A variant recombined efficiently with hemin at room temperature. Figure 2A presents a portion of the downfield-shifted hyperfine region of I12A ferricyt b5 and compares it to the wild-type spectrum (Figure 2D). In this chemical shift window and beyond, the two spectra resemble each other closely, indicating that the axial coordination of the heme group and the structure of the heme pocket are conserved. Assignments of the heme signals in the I12A ferricyt b5 spectrum were transferred from the wild-type protein and verified with homonuclear experiments. A portion of the two-dimensional nuclear Overhauser effect spectroscopy (NOESY) data is shown in Figure S1 of the Supplementary Information, and a selected set of chemical shifts is listed in Table I. Integration of several resonances in Figure 2A returned a heme orientational isomer ratio A:B of 1.6:1, within an error of that of the wild-type protein.35 A relatively large difference in chemical shift between the two proteins was observed for the 8-CH3 signal of the minor heme orientational isomer (at ˜24 ppm). The resolution of this pair of peaks and the partial resolution of the 2-α-vinyl resonances of the major isomer were useful in the heme-transfer experiments described later. Signals from the 6- and 7-propionate substituents were the most strongly affected, an observation to which we will return.

Downfield-shifted hyperfine region of ferricyt b5. (A–D) The holoprotein of the I12A variant was mixed with wild-type apoprotein to measure the heme exchange rate. (A) Spectrum of the mixture after 8 min of equilibration; the major species is the holoprotein of the I12A variant. Heme signals are a, 3-CH3 of the minor heme orientational isomer (“B” isomer); b, 2-α vinyl of the major heme orientational isomer (“A” isomer); c, 8-CH3 of isomer B; d, 3-CH3 of isomer B; e, 7-α propionate of isomer B; f, 6-α propionate of isomer B. (B) Spectrum of the mixture after 71 min of equilibration. (C) Spectrum of the mixture after 155 min of equilibration. (D) Spectrum of the mixture after 12 h of equilibration; the major species is the wild-type holoprotein. (E) Spectrum of the N57P variant.
| Assignment | WTb | N57P | I12A | D60R | PE |
|---|---|---|---|---|---|
| 1-CH3 | 10.73 | 10.72 | 10.68 | 10.31 | 8.12 |
| 3-CH3 | 14.44 | 14.25 | 14.55 | 14.51 | 16.18 |
| 5-CH3 | 20.42 | 20.52 | 20.33 | 20.01 | 16.36 |
| 8-CH3 | 2.90 | 2.65 | 2.85 | 3.00 | 3.03 |
| α-meso | −2.98 | −3.13 | −3.05 | −3.00 | −3.87 |
| β-meso | 9.29 | 9.25 | |||
| γ-meso | −0.37 | −0.36 | −0.55 | −0.49 | |
| δ-meso | 10.19 | 10.03 | 10.08 | ||
| 2-Hα | 27.43 | 27.06 | 27.50 | 27.54 | 24.86 |
| 2-Hβc | −7.00 | −7.13 | −7.14 | −7.01 | −6.94 |
| 2-Hβt | −7.37 | −7.37 | −7.53 | −7.47 | −7.18 |
| 4-Hα | 5.10 | 5.01 | |||
| 4-Hβs | 2.57 | 2.54 | |||
| 6-Hα | 14.98 | 15.13 | 14.99 | 14.79 | 14.27 |
| 6-Hα′ | 14.87 | 15.04 | 14.95 | 14.63 | 14.27 |
| 6-Hβ | −0.96 | −1.03 | −0.99 | −0.98 | −1.53 |
| 6-Hβ′ | −1.33 | −1.77 | −1.82 | −1.74 | −1.85 |
| 7-Hα | 19.59 | 19.60 | 19.52 | 19.74 | 20.60 |
| 7-Hα′ | −1.76 | −1.64 | −1.40 | −1.18 | −0.94 |
| 7-Hβ | 1.69 | 1.68 | 1.63 | 1.70 | 1.55 |
| 7-Hβ′ | −3.76 | −3.82 | −3.82 | −3.76 | −3.91 |
| His39 HN | 9.01c | 8.98 | 8.98 | 8.99 | |
| His39 Hα | 6.90 | 6.87 | 6.92 | 6.94 | 7.4 |
| His39 Hβ | 16.33 | 16.42 | 16.48 | 16.50 | 16.48 |
| His39 Hβ′ | 7.37 | 7.34 | 7.42 | 7.39 | 7.14 |
| His63 HN | 11.11c | 11.09 | 11.09 | 11.17 | |
| His63 Hα | 7.45 | 7.44 | 7.44 | 7.48 | |
| His63 Hβ | 10.18 | 10.54 | 10.22 | 10.22 | |
| His63 Hβ′ | 9.95 | 9.56 | 9.95 | 10.01 |
In addition to the heme signals shown in Figures 2A and S1, it was possible to assign a large number of resonances with uniformly 15N-labeled protein. A 1H-15N heteronuclear single-quantum coherence (HSQC) spectrum is shown in Figure 3, which can be compared with that published by Guiles and coworkers.37 The cross peaks are labeled according to their origin, with A and B denoting the two heme orientations where possible. The side chains bordering the heme group gave rise to NOEs typical of the wild-type geometry, for example in the major isomer: Leu25 and Ala54 to 3-CH3, Phe35 to 1-CH3, Pro40 to 7-propionate, Val45 to 5-CH3, among others (Figure S1). The signals from Ala12 were readily recognized amidst resonances that matched well those in the wild-type protein. The three-dimensional data allowed for the unambiguous assignment of strong NHi-NHi+1 NOEs throughout H1, in agreement with the presence at position 12 of a residue that favored α structure. Figure 4 displays the corresponding region of the homonuclear data.

1H-15N HSQC spectrum of I12A ferricyt b5 at pH 7.2 and 25°C.

Portion of a NOESY spectrum of I12A ferricyt b5 at pH 7.2 and 25°C. The sequential NOEs of H1 (Leu9–Lys14) are indicated with a dashed line. The NH of Leu9 is at 9.50 ppm and that of Lys14 at 7.37 ppm.
Ala12 had a number of nonsequential dipolar contacts, a few of which are shown in Figure 5. Other NOEs involving side chains of hydrophobic core 2 (for example, Trp22–His15, Trp22–Ile76, Tyr7–Ile76, and Tyr7–Val29, as shown in Figure S2 of the Supplementary Information) suggested a largely preserved three-dimensional structure. The I12A replacement caused chemical shift deviations larger than 0.15 ppm for some protons in Thr8, Leu9, Lys16, Val23, Ile24, Leu25, Gly52, Ala54, and Ile76 in both the A and B isomers. These residues form a cluster extending from Ile12 to the α-meso edge of the heme group. In contrast, residues 32–49 and 65–74 were the least affected. Although the chemical shift changes demonstrated widespread readjustments of the I12A holoprotein structure, the heme binding energy was sufficient to offset the cost of creating a cavity in hydrophobic core 2 and to organize the protein into the wild-type conformation.

Portion of a NOESY spectrum of I12A ferricyt b5 at pH 7.2 and 25°C. The chemical shift of Ala12 CβH3 is indicated by a vertical dashed line. The NOEs of this group are labeled on the side. The weak effects to the ring of Trp22 suggest a conserved geometry of core 2.
The N57P variant also bound hemin specifically. Figure 2E contains a portion of the downfield-shifted hyperfine region of N57P ferricyt b5. Despite proximity of Asn57 to the 4-vinyl group, the A:B ratio in N57P holocyt b5 was within error of that in the wild-type protein. The changes in heme chemical shifts were more pronounced than in the I12A variant (excluding the propionate shifts, Table I), but remained small. Compared to the rat wild-type (A isomer), the signals from the 3-CH3 and 8-CH3 shifted upfield, whereas those from the 1-CH3 and 5-CH3 shifted downfield. A stick representation of the methyl shifts is shown in Figure 6, which illustrates the magnitude of the deviations relative to three wild-type proteins, from rat, beef, and chicken microsomes. The lines of N57P ferricyt b5 are sharper and closer to the wild-type chemical shift than those of the PE protein.22

Diagram representing the chemical shift of heme methyls and meso protons in various proteins. Heme methyls (labeled 1, 3, 5, 8) are in filled symbols. Meso protons (α, β, γ, δ) are in open symbols. Chicken, beef and rat refer to the wild-type shifts determined by La Mar and coworkers7; t-beef (trypsin-solubilized) and V45E refer to the shifts determined by Huang, Wu and coworkers for the bovine protein.38 Other proteins are the variants of rat cyt b5 as described in the text.
Two-dimensional homonuclear data were used to assign protein resonances in the N57P variant. Signals in the Ala54–His63 stretch were identified; H4 and the Val61–His63 turn appeared to be stably formed. The same typical NOEs listed earlier were detected, e.g., Leu25 and Ala54 to 3-CH3, Phe35 to 1-CH3, Pro40 to 7-propionate, and Val45 to 5-CH3. Unlike the I12A substitution, the N57P replacement resulted in protein chemical shift perturbations confined to the environment of the change, i.e., strand β5 (between H3 and H4) and H4. H1 was relatively unaffected.
The HAY variant contained two residues capable of axial ligation on the 39 side of the heme group (His35 and Tyr46). Hemin binding resulted in an optical spectrum different from that of H39A ferricyt b5, which at neutral pH is typical of a His–H2O iron ligation (not shown). The Soret maximum of the HAY apoprotein mixed with substoichiometric amounts of hemin was 410 nm; Q bands were observed at 561 nm and 530 nm, as well as a charge transfer shoulder at 630 nm (Figure S3). These features indicated both low- and high-spin contributions to the electronic structure, with only moderate resemblance to the spectrum of holo HasA.39 Moreover, the 1H NMR spectrum did not contain resolved hyperfine shifted signals, which suggested nonspecific binding or largely enhanced heme pocket dynamics. Further characterization will be necessary to ascertain the participation of His35 and Tyr46 in HAY cyt b5 heme coordination, but it appeared that the energy involved in the reconformation of the heme pocket at neutral pH was too high to be compensated for by a stable alternative ligation scheme.
For the variants that bind heme specifically, the heme and axial ligand hyperfine shifts are sensitive reporters of local structure.5, 35 For example, heme methyl signals can be used as indicators of the orientation of the axial histidine imidazole planes with respect to each other and to the heme plane.40 As shown in Figure 6, the shifts caused by the sequence changes in rat cyt b5 were small. Except for the PE variant, the magnitude and pattern were consistent with a minimal rotation of the axial histidines, likely because of H4 distortions (N57P and D60R) or a slight readjustment of the heme in its cavity (I12A). The PE variant displayed chemical shift deviations comparable in magnitude to those observed between rat and chicken ferricyt b55 and to those reported in the V45E variant of bovine ferricyt b5.38 Val45 is located in H3 directly above the β-meso proton in the A isomer. Its replacement causes well-described distortions of the heme cavity structure.41 The PE chemical shift data were consistent with <5° variations of the angle between the imidazole planes (β) and the angle between the bisector of these planes and the heme NA-NC axis (φ); complete chemical shift information would be required to determine the axial ligand geometry to a higher accuracy.
Effects of the Replacements on the Holoprotein—Stability
Figure 7 illustrates the thermal denaturation of I12A ferricyt b5. The process was not fully reversible as typical for b hemoproteins, and slight variations in the beginning of the transition were observed from sample to sample. Nevertheless, each curve could be modeled with a two-state equation of denaturation. The midpoint of the transition (Tm) was a consistent 60.5°C ± 0.4°C, which was used as a measure of stability. Under the same condition, the wild-type protein had a midpoint of 70.0°C ± 0.1°C; values for other variants are listed in Table II. On a Tm scale of stability, the I12A variant was slightly higher than the PE variant and lower than the P81A protein.

Thermal denaturation of ferricyt b5 monitored by the change in absorbance at the Soret maximum. The curves represent the apparent fraction of unfolded protein as a function of temperature for the I12A variant (open squares), the wild-type protein (open diamonds), and the N57P variant (open circles). The midpoints are listed in Table II.
| Protein | Holo | Apo | Holo | ||||
|---|---|---|---|---|---|---|---|
| Tma | ΔS°b | Tma | ΔG°c | md | ΔG°e | ΔΔG°f | |
| WT | 70.0 ± 0.1 | 1.0 | 46.1 ± 0.6 | 6.4 ± 0.4 | 3.9 ± 0.1 | 27.5 ± 0.8 | 0.0 |
| I12A | 60.5 ± 0.4 | 0.9 | ∼31 | ∼2 | (3.7 ± 0.8) | (17 ± 3) | (−9.5) |
| N57P | 73.6 ± 0.1 | 1.2 | 45.1 ± 0.3 | 5.9 ± 0.4 | –g | >27.5g | |
| PE | 58.0 ± 0.1 | 0.9 | 44.5 ± 0.3 | 6.5 ± 0.5 | 4.4 ± 0.2 | 20.0 ± 0.8 | −11.5 |
| P81A | 63.3 ± 0.1 | 1.0 | 25.7 ± 0.7 | 0.1 ± 0.1 | 4.4 ± 0.2 | 22.7 ± 0.8 | −8.0 |
| D60R | 76.0 ± 0.1 | 1.0 | 46.9 ± 0.8 | 7.0 ± 0.6 | –g | >27.5g | |
- Results from this work are marked in italics.
- a Midpoint of the thermal denaturation, in degree Celsius (20 mM phosphate, pH 7.2).22
- b In kJ mol−1 K−1 at Tm.22
- c In kJ mol−1, at 25°C, from the thermal denaturation data with ΔCp fixed at 4.2 kJ mol−1 K−1, except for I12A apocyt b5, which was given a value of 0 (see text).
- d In kJ mol−1 M−1.
- e By chemical denaturation, in kJ mol−1, at 25°C, and in the absence of urea.22
- f In kJ mol−1 M−1, at 25°C, at 7M urea and with respect to the wild-type value.
- g The data did not allow for an accurate determination of the unfolded baseline. Values in parentheses are marginally reliable and were not used in Figure 12B.
Urea denaturation of I12A holocyt b5 was monitored by far-UV circular dichroism (CD). Although the curves (not shown) returned a consistent midpoint concentration of ˜4.5M, the slope in the transition varied (m = 3.7 ± 0.8 kJ mol−1 M−1), again mostly because of discrepancies in the pre- and early denaturation region, and led to a large uncertainty in the estimated ΔG° in the absence of urea (17 ± 3 kJ mol−1). We have noted that despite irreversibility, the ΔG° (25°C) obtained from thermal curves matched well the values obtained by chemical denaturation.22 The I12A variant, however, did not seem to follow this trend.
Bertini and coworkers have inspected the consequences of exposing wild-type cyt b5 to 2M guanidinium chloride.5 They observed two stretches of fragile structure: 33–38 (H2 leading to the axial His39) and 62–64 (turn between H4 and H5, containing the axial His63). The tendency of H2 to unravel first in the holoprotein contrasts with its behavior in the apoprotein, where it is a stably folded element of secondary structure.19 The mechanisms of destabilization by chemical denaturation and by I12A replacement are clearly different. They may, however, reveal similar weaknesses in the structure. It is noteworthy that the heme propionate side chains, located close to Gly62, experience comparatively large Δδs regardless of the ΔΔG° associated with the replacement. Thus, the data presented here support that this region of the protein is prone to rearrangement, whereas H2 is not particularly affected by Type I substitutions.
The N57P variant had a higher Tm than the wild-type protein (73.6°C ± 0.1°C), although the replacement was expected to interfere with the proper folding of H4 and destabilize the heme–apoprotein complex. The detection of NHi-NHi+1 and side chain NOEs before and after Pro57 and the constant A:B isomer ratio suggest that the modified β5–H4 structure packs efficiently against the heme group, and it is difficult to relate the change in stability to specific structural features of the native state.
Effects of the Replacements on the Apoprotein—Structure
The I12A replacement had a marked effect on the apoprotein properties. Figure 8A displays the corresponding 1H NMR spectrum. Figure 8C presents the N57P apoprotein spectrum, which is directly comparable to that of the wild-type protein. Relative line intensities in the I12A apocyt b5 spectrum show that, at room temperature, the folded state is only partially populated. The methyl group near −1 ppm arises from Ile76 CδH3 (Figure 1B) and its upfield shift, caused by Trp22, is a good indicator of the integrity of core 2.42 Ile76 line broadening and reduced ring current shift suggested enhanced structural fluctuations and a lowering of the activation energy barrier separating the native conformation from others. Figure 9 illustrates the changes upon cooling a sample of I12A apoprotein. As the temperature was lowered, resolved signals grew in intensity, demonstrating that the protein could be refolded. The data suggested that some structural information might be obtainable at low temperature, although complicated by residual unfolded state population and a broad range of exchange rates.

One-dimensional 1H NMR spectra of apocyt b5 at 25°C in 90% 1H2O/10% 2H2O, pH 7.2–7.3. (A) I12A variant; (B) HAY variant; (C) N57P variant. The vertical scaling is arbitrary.

One-dimensional 1H NMR spectra of I12A apocyt b5 in 90% 1H2O/10% 2H2O, pH 7.2, as a function of temperature. (A): 25°C; (B), 20°C; (C), 15°C, (D), 10°C; (E), 5°C. The difference between the A traces in this Figure and Figure 8 reflects the difficulty in generating consistent samples of apoprotein in the thermal transition.
Among the residues that were assigned by 2D and 3D methods in the conformer resembling most the native wild-type apocytochrome were His15, Trp22, Val29, and Ile76. Partial assignments were also obtained for Leu9, Ile24, and Leu79. The formation of β structure was evident in the fingerprint region (Figure S4 of the Supplementary Information). H1 could be traced; its state was not well defined as the signals of Ala12 and Gln13 overlapped. These fragmentary data supported that the sequence still coded for the same specific tertiary structure as the wild-type sequence and illustrated the decoupling of stability and specificity in this system.43
In contrast to the I12A apoprotein, the HAY apoprotein displayed a higher proportion of native form at room temperature (Figure 8B). The position of the lines and their width also indicated dynamic properties closer to those of the wild-type protein. Upon cooling, signals typical of the β structure of cyt b5 emerged between 5 and 6.5 ppm (Figure S5), including the CαH of Trp22,44 which was not detectable at room temperature despite the wild-type like proximity of Ile76 to the indole ring. Two-dimensional homonuclear data at low temperature confirmed the formation of several of the core-2 contacts listed earlier. The totally correlated two-dimensional spectroscopy (TOCSY) data also revealed exchange cross peaks supporting slow interconversion processes. Comparatively larger chemical shift deviations were observed throughout the structure, which were likely to be caused by intermediate conformational exchange among distinct states as well as structural distortions of the limiting conformation compared to the native wild-type geometry.
Effects of the Replacements on the Apoprotein—Stability
The thermal denaturation of the apoproteins was followed by absorbance or CD measurements. The fit for the N57P variant data was performed as reported previously,22 with a two-state model of unfolding and a ΔCp fixed at 4.2 kJ mol−1 K−1. When the fit was repeated with different ΔCp values, χ2 exhibited a broad minimum within 3.6–4.2 kJ mol−1 K−1. Variations in this range returned comparable Tms and ΔH°s. Overall, the N57P replacement had no effect on the thermal denaturation of the apoprotein (Table II).
The A284 curve obtained for the I12A variant was considerably shallower than that for the wild-type protein, and the difference in signal between folded and unfolded state was reduced (Figure 10A). This latter feature was attributed to the perturbation of the environment of Trp22, likely through the formation of a cavity invaded by solvent. Although the wild-type and N57P curves were suitable for analysis, the curve for the I12A variant, which lacked sufficient definition, could not be reliably fitted. When ΔCp was varied as for the N57P variant, a broad minimum of χ2 was found near 0 kJ mol−1 K−1. With ΔCp = 0, the Tm was ˜31°C, and the protein was ˜70% native at 25°C. The thermal transition of I12A apocyt b5 was also monitored by CD. Figure 10B presents a plot of ellipticity that confirmed reduced cooperativity and the low stability of the secondary structure of the protein observed by NMR spectroscopy. In addition, thermal unfolding was not completely reversible unlike that of the wild-type and N57P proteins. The HAY apoprotein was also unstable and, like the I12A apoprotein, did not yield interpretable curves. Under the same denaturation conditions, the transition was reversible to a lesser extent than for the I12A apoprotein.

(A) Thermal denaturation of apocyt b5 monitored by absorbance measurements at 284 nm. Curves for the wild-type protein (diamonds) and the I12A variant (squares) are shown. The data were normalized to match the absorbance in the denatured state. (B) Thermal denaturation of apocyt b5 monitored by circular dichroism at 221 nm. The ellipticity of a ˜7 μM solution of I12A variant is plotted versus temperature.
Heme Transfer Experiment
In view of the I12A cyt b5 stability data, only one heme transfer experiment was practical: from I12A holoprotein to wild-type apoprotein. The kinetics of transfer of the heme group is summarized in Figure 11. Shown are the profiles for the major and minor forms of I12A cyt b5 and the major and minor forms of the wild-type protein. Fitting the data with a single exponential decay returned the rate constants for heme loss from the variant: (7.3 ± 0.1) × 10−3 min−1 for the A isomer and (12.4 ± 0.1) × 10−3 min−1 for the B isomer. These constants were in a 1.7:1 ratio, in close agreement with the A:B equilibrium ratio. The figure also shows that in the early stage of transfer, the two isomers of the acceptor protein were equally populated.

Kinetics of heme transfer from the I12A variant to the wild-type protein. The vertical scale is normalized to heme concentration in mM. The curves represent the decay of the major (squares) and minor (triangles) heme orientational isomer of the I12A protein and the recovery of the major (circles) and minor (diamonds) heme orientational isomer of the wild-type protein. Spectra collected during the exchange and used for the concentration evaluation are shown in Figures 2A–2D.
The total variant and wild-type population (sum of the minor and major isomer concentrations) can be fit to obtain an apparent rate constant for heme loss for comparison with optical data. Again, a single exponential fit accounted reasonably well for the trend. The value was k−H = 9.1 × 10−3 min−1, which can be compared to data previously reported for the wild-type protein and the destabilized PE variant. The wild-type protein has a dissociation rate constant of ˜1.0 × 10−3 min−1 obtained by transfer to apomyoglobin,28 significantly slower than that of the PE variant (25 × 10−3 min−1).22 On the assumption that the heme on-rate constants were unchanged by the sequence alterations (as observed in myoglobin),45 the ratio of these off-rate constants scaled directly with heme affinity and was converted into a ΔΔG° value. Binding of ferric heme by the I12A variant is 2.5 kJ mol−1 more favorable than by the PE variant and 5.5 kJ mol−1 less favorable than by the wild-type protein. Unlike in the PE variant, which intentionally perturbed the heme site, the I12A variant displayed an unchanged structure around the heme group; the effect can be attributed to energy spent in refolding the apoprotein or in nonspecific binding. A 5.5 kJ mol−1 difference in stability compared to the wild-type protein is plausible in the holoprotein state, but may be an overestimate in the apoprotein state. Experimental evidence reported by Ihara et al.46 on H39L and H63L cyt b5 indicates that heme association and dissociation rates are related to loop conformational change. It is possible that, in the I12A variant, the rate constant for heme association was also affected by the apoprotein refolding step, in which case the free energy corresponding to the affinity should be adjusted. The fact that native and unfolded states of the I12A variant were in slow exchange on the NMR time scale indicated additional complication for data interpretation.
Relationship to Other Cytochrome b5 Data
We first consider briefly HAY cyt b5. The axial His39 and His63 do not play equivalent roles in the wild-type holoprotein. Despite evidence for stronger His39–heme than His63–heme interactions,47 cyt b5 lacking His39 loses heme at a slower rate than that lacking His63.46 We also observed that H39A ferricyt b5 is more stable than H63A ferricyt b5.19 Thus, replacing His39 in making the HAY variant left open the possibility that the heme would bind specifically to His63. Phe35 is in contact with the heme group, and its replacement with histidine in the bovine variant diminishes considerably the stability of the holoprotein at pH 7.13 Leu46 is in contact with the heme group, and it is possible that its replacement is disruptive as well. A tyrosine at position 46 is found in the cyt b5 domain of several steroid-binding proteins,48 but this provides little insight since these proteins may not bind heme tightly.49 Regardless of the consequences of individual substitutions, their combination was detrimental to the stability of the cytochrome.
Many artificial variants of bovine cyt b5 have been studied for their structure, stability, and heme iron redox potential. Tm values and estimates of the change in stability of the holoprotein caused by the sequence changes are available for several of these variants. Tm and ΔG° data sets, when compiled for the denaturation of well-behaved single domain proteins devoid of cofactors, are expected in a first approximation to exhibit a linear trend between ΔΔG° and ΔTm, with a slope representing an averaged ΔS° parameter.50 For this purpose, the wild-type Tm is a convenient reference temperature, and the ΔΔG° values at that temperature are represented as ΔΔG° (50%). This approach circumvents in part the difficulty in obtaining accurate ΔCp values. For the cytochromes, the ΔΔG° values extracted from thermal denaturation monitored by optical means are suspect because of nonreversibility. Tm, in contrast, is largely impervious to the changes in thermodynamic parameters used for fitting as long as the native and unfolded baselines are well defined. As an alternative to the thermal ΔG° values, urea denaturation ΔG° values can be used. To avoid the long linear extrapolation of the data necessary to determine ΔG° in the absence of urea, ΔG° values at urea concentrations in the transition are acceptable. As for the thermal data, a convenient reference is the urea concentration that yields ΔG° = 0 for the wild-type protein (7M). Table II lists the urea ΔΔG° (50%) obtained for PE at 25°C and P81A cyt b5 from data collected previously.22
Figure 12A presents the thermal ΔΔG° (50%) versus ΔTm plot for the bovine holoprotein data published by others. The sites targeted in this sequence were Val4551 and Val6152 located in the disordered region of the rat apoprotein, and Phe35,13 in the folded H2. These data defined a straight line (R2 = 1.0). The slope, which was obtained without the wild-type points at (0,0), was 1.1 kJ mol−1 K−1, in agreement with the reported apparent average ΔS° for the bovine proteins.52 The urea ΔΔG° (50%) values (slope of 0.9 kJ mol−1 K−1 with R2 = 0.90) were collected at different temperatures and were therefore not considered quantitatively. The four urea ΔΔG° (50%) data points from the rat proteins (wild-type, PE, I12A, and P81A) are indicated on the plot with an estimate of experimental error. They fell near the best line defined by the bovine data. The ΔS° at Tm obtained from the thermal denaturation of the rat proteins was ˜1.0 kJ mol−1 K−1 (Table II). Thus, even though the thermal process did not fulfill the criteria required for thermodynamic parameter determination, this approach led to self-consistent results. The correlation deteriorated when using ΔΔG° values extrapolated to buffer. This was due to the different slopes of ΔG° as a function of urea concentration exhibited by the variants.

(A) Plot of ΔΔG° (50%) versus ΔTm for bovine and rat ferricyt b5. The squares represent published thermal data on F35, V45, and V61 variants of the bovine cytochrome. In each case, the ΔΔG° and ΔTm values were obtained by reference to the wild-type protein; points for the wild-type are at (0,0) and are not shown. The four circles represent rat ferricyt b5 data (urea, Table II). (B) Plot of ΔG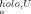 versus ΔG
versus ΔG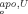 and predictions from Eq. (1) as described in the text. Calculated behaviors are given by line a (rat wild-type), line b (bovine wild-type), line c (I12A variant), and line d (PE variant). The points represent experimental ΔG° values (see text). The circle on line b represents bovine wild-type cyt b5.12
and predictions from Eq. (1) as described in the text. Calculated behaviors are given by line a (rat wild-type), line b (bovine wild-type), line c (I12A variant), and line d (PE variant). The points represent experimental ΔG° values (see text). The circle on line b represents bovine wild-type cyt b5.12
The final state achieved upon holoprotein denaturation is not well defined. Under conditions of heat energy or urea concentration defining the early stage of the unfolded baseline, it is likely that a fraction of the protein population contains nonspecifically bound heme. This fraction is capable of refolding to the native holoprotein state in a unimolecular process. We have observed that the apparent ΔG° values obtained from the thermal denaturation match those obtained by urea denaturation for certain proteins.22 This observation is not expected to hold for all replacements,53 as demonstrated by the I12A variant. Nevertheless, the correlation between ΔTm and apparent urea ΔΔG° (50%) is noteworthy and the plot in Figure 12A provides a phenomenological relationship that can be used to anticipate or examine the change in stability corresponding to an observed ΔTm.
According to the linear ΔΔG° versus ΔTm relationship, the Tm of the N57P variant corresponded to an increase in stability of 3 kJ mol−1 at 7M urea. The effect of proline introduction on protein stability was recently reviewed by Varadarajan and coworkers.53 The entropic contribution to the free energy of the unfolded state was originally proposed to be ˜5 kJ mol−1 at room temperature,54 but may be larger. If such is the case for the N57P variant, unfavorable interactions in the native state would have to reduce the entropic impact to lower Tm and ΔG°.
Trends that involve holoprotein and apoprotein properties were not readily extracted from the data. Equation (1) predicts a behavior that can be tested, provided some assumptions are made. The first step is to estimate the nonspecific heme binding constant for the wild-type rat holoprotein. Nonspecific interactions include ligation bonds with histidines other than the axial ones and contacts with exposed hydrophobic side chains. A value of 3.8 × 10−13 M was used for K−H; this is derived from association and dissociation rate constants.28, 55 K−H, along with measured Kholo,U and Kapo,U, led to a K−NS of 1.6 × 10−8 M at 25°C (ΔG° ≈ 44 kJ mol−1), which is in line with expectations based on other hemoproteins.27, 56 In a second step, K−NS was taken as independent of the protein, and changes in heme affinity were attributed solely to a change in the rate constant for heme loss. Thus, K−H/K−NS could be evaluated for two of the rat cytochrome variants. The relationship between the thermal ΔG°holo,U and ΔG°apo,U predicted by Eq. (1) is represented in Figure 12B. Line a was calculated with the wild-type parameters determined earlier. Line b was calculated with similar parameters from published wild-type bovine data.14, 51, 52, 55 Lines c and d used the rate constants of heme loss from the I12A variant and PE variant, respectively. As can be seen, ΔG°holo,U is independent of ΔG°apo,U as long as the apoprotein stability is above 6 kJ mol−1. This corresponds to Kapo,U < 0.08 for use in Eq. (1).
Figure 12B also contains experimental data points, which are few because of the limited availability of complete holo- and apoprotein data sets. The rat and bovine wild-type points, whose ΔG°holo,U and ΔG°apo,U were used to estimate K−NS, naturally fell on lines a and b, respectively. The D60R cyt b5 data point is above the wild-type line because of slightly higher holoprotein stability. The PE cyt b5 data point was above its calculated value (line d), at a level corresponding to a 2.5-fold deceleration of heme loss. The P81A data point lied in the region of the plot where the apoprotein stability influences the apparent holoprotein unfolding. The predicted rate of heme loss that goes through the point is ˜1.5 times faster than wild-type. The P81A variant has a measured rate of heme loss between those of the wild-type and PE proteins,22 possibly consistent with the derived 1.5 factor. The I12A data point is likely to lie above line c. Overall, the variant data suggested that the rate of heme loss estimated from the heme transfer experiments was not the sole determinant of relative affinity.
It is possible to stabilize the bovine apoprotein significantly (an extra 7 kJ mol−1) by reinforcing interactions in core 2. The H15R/S20E double replacement in bovine microsomal cyt b5 accomplishes this,12 presumably by installing a salt bridge stabilizing the turn between H1 and β4. This has a comparatively small effect on the holoprotein denaturation. As the stability of the wild-type protein is higher than that of the rat isoform (11 kJ mol−1), Eq. (1) predicts no contribution of the apoprotein thermodynamic properties to those of the holoprotein. The data point for this Type II replacement is predicted to fall on line b.
CONCLUSIONS
When destabilizing replacements such as I12A and HAY are made to apocyt b5, the polypeptide chain is still capable of accessing a conformation resembling that of the wild-type protein. The substitutions are expected to raise the free energy of this state, but even in the most severe case, the native state remains separated from other conformations by an activation energy barrier sufficient to cause slow exchange on the chemical shift time scale. When replacements such as PE, N57P, and D60R are made in the region that has a low intrinsic probability of forming stable structure, the apoprotein does not appear to be perturbed. The response of the holoprotein varies because of the adaptability conveyed by this region. For example, the N57P variant cannot adopt a geometry identical to that of the wild-type protein near the heme group, but it experiences an increase in stability likely due to a combination of compensating adjustments in the native state and unfolded state effects. Although denaturation of b hemoproteins monitored by optical methods often yields marginally reliable thermodynamic data, useful correlations were derived. The PE, N57P, and D60R variants illustrate the decoupling of holoprotein and apoprotein stability, whereas the P81A, I12A and HAY variants reveal a different aspect of the energetic landscape. The P81A and I12A cyt b5 have a low apoprotein stability reflected in the holoprotein stability, whereas HAY cyt b5 has somewhat reduced apoprotein stability and impaired heme binding, exposing the limits of the ability of the disordered region to refold. Irreversibility of the I12A and HAY apoprotein denaturation also suggests the participation of the disordered loop in establishing solubility properties.
MATERIALS AND METHODS
Protein Expression and Purification
Rat microsomal cyt b5 genes with the desired mutations (N57P, I12A, and F35H/H39A/L46Y) were prepared by polymerase chain reaction (QuickChange, Stratagene, La Jolla, CA) as previously described for other mutants.22 The sequence of each mutated gene was verified at the Penn State Nucleic Acid Facility. Transformed BL21 DE3 E. coli cells were grown in M9 minimal medium containing ampicillin, and protein expression was induced with isopropyl β-thiogalactoside. Cell lysis was achieved by ultrasonication after resuspension in buffer containing phenylmethylsulphonyl fluoride (1 mM). For the N57P variant, a solution of hemin chloride was added to the supernatant, and the protein was purified in the holoprotein state. The I12A and HAY variants were purified in the apoprotein state. Further purifications steps were as applied to other heme-reconstituted variants.20, 22 Whereas the holoprotein of the I12A variant could be obtained by hemin addition, the HAY variant, which proved to be unstable and more difficult to handle, was maintained in the apoprotein state. Uniformly 15N-labeled I12A protein was prepared in the same fashion as the unlabeled protein, with M9 medium containing 15N- ammonium sulfate.
Ferric holoprotein concentrations were determined by using an extinction coefficient (ε) of 130 mM−1 cm−1 at the Soret maximum (˜412 nm) for all holoproteins.57 I12A apocyt b5 concentration determinations used an ε value of 10.6 mM−1 cm−1 at 280 nm as for the wild-type apoprotein.58 This value only gave an estimate because this variant was expected to present structural distortions near Trp22. A value of 12.9 mM−1 cm−1 was used for HAY apocyt b5 as per the composition of the protein.
Thermal Denaturation
Apoprotein and ferric holoprotein samples were prepared in 20 mM phosphate buffer at pH 7.2, at concentrations ≤100 μM or ≤ 10 μM, respectively. UV–visible data were collected on an AVIV model 14 DS spectrophotometer; the sample temperature was controlled with a thermoelectric device. Spectra (440 nm to 260 nm) were obtained in 2°C steps with an equilibration period of 3 min. Reversibility was 95–100% for apoproteins, except the I12A and HAY variants, and 70% and below for the holoproteins. Apoprotein data were analyzed at 284 nm and holoprotein data at 413 nm. Apoprotein CD data were also collected on a JASCO-810 system with the same schedule of heating and cooling. The wavelength was 221 nm and signal was averaged for 4 s.
Thermal denaturation data were treated with a two-state model of denaturation according to a modified Gibbs–Helmholtz equation accounting for a linear variation of the optical properties of the native and unfolded states.59 The thermal transitions were fitted with a fixed ΔCp identical to that of the wild-type holoprotein (6.0 kJ mol−1 K−1) or the wild-type apoprotein (4.2 kJ mol−1 K−1).60 As holoprotein transitions are reproducible and not fully reversible, Tm is the only fitted parameter used to qualify stability. Data for the I12A and HAY apoproteins were not fitted for lack of well-defined baselines.
Chemical Denaturation
Urea denaturation experiments of I12A ferricyt b5 were performed at 25°C as described previously.22 The protein concentration was 10–25 μM in 20 mM phosphate buffer (pH ˜7.3). Equilibration time at each concentration was 5 min with stirring. Far-UV CD spectra were collected from 210 to 250 nm with the spectropolarimeter mentioned earlier. Data analysis was performed using the ellipticity at 222 or 223 nm and the program SAVUKA.61 A linear free energy relation and a two-state equation were used to fit the data.59
NMR Spectroscopy
NMR data were collected on a Bruker DRX-600 spectrometer (14.1 T, operating at a 1H frequency of 600.05 or 600.18 MHz). Sample concentrations were 1–2 mM in 20 mM phosphate buffer or in 2H2O, pH 7.2–7.4, 25°C. Two-dimensional NMR experiments (double-quantum-filtered correlated spectroscopy, relaxation compensated TOCSY, and NOESY) and 15N-separated NOESY and TOCSY data were performed as in past work.62 Data were processed with XWIN-NMR (Bruker) or NMRPipe63 and analyzed with XWIN-NMR (Bruker) or Sparky.64
Heme Transfer
These were performed as described previously22 with approximate initial concentrations of 0.7 mM I12A holoprotein and 3.0 mM wild-type apoprotein. Solutions of the I12A holoprotein and excess wild-type apoprotein were mixed in the NMR tube, and one-dimensional data were collected as a function of time. Signals from the major and minor isomers of the holoproteins (Figure 2) were simulated with DMFit.65
Acknowledgements
The authors thank Kristen Yarmey for assistance with the preparation of the N57P variant; Daniel Landfried for assistance with denaturation experiments; Dr. David Vuletich for assistance with NMR data collection; Drs. Jane Knappenberger and Christopher Falzone for useful discussions; and Dr. Ronald Davis for careful reading ofthe manuscript. Figures 1B and 1C were prepared with MOLSCRIPT.66

