

Factors governing 310-helix vs α-helix formation in peptides: Percentage of Cα-tetrasubstituted α-amino acid residues and sequence dependence
Abstract
As an additional step toward the dissection of the factors responsible for the onset of 310-helix vs α-helix in peptides, in this paper we describe the results of a three-dimensional (3D) structural analysis by x-ray diffraction of the Nα-acylated heptapeptide alkylamide mBrBz–L-Iva–L-(αMe)Val–L-Abu–L-(αMe)Val–L-(αMe)Phe–L-(αMe)Val–L-Iva–NHMe characterized by a single (L-Abu3) Cα-trisubstituted and six Cα-tetrasubstituted α-amino acids. We find that in the crystal state this peptide is folded in a mixed helical structure with short elements of 310-helix at either terminus and a central region of α-helix. This finding, taken together with the published NMR and x-ray diffraction data on the all Cα-methylated parent sequence and its L-Val2 analog (also the latter heptapeptide has a single Cα-trisubstituted α-amino acid) strongly supports the view that one Cα-trisubstituted α-amino acid inserted near the N-terminus of an Nα-acylated heptapeptide alkylamide sequence may be enough to switch a regular 310-helix into an essentially α-helical conformation. As a corollary of this work, the x-ray diffraction structure of the Nα-protected, C-terminal tetrapeptide alkylamide Z–L-(αMe)Val–L-(αMe)Phe–L-(αMe)Val–L-Iva–NHMe, also reported here, is clearly indicative of the preference of this fully Cα-methylated, short peptide for the 310-helix. As the same terminally blocked sequence is mixed 310/α-helical in the L-Abu3 heptapeptide amide but regular 310-helical in the tetrapeptide amide and in the parent heptapeptide amide, these results point to an evident plasticity even of a fully Cα-methylated short peptide. © 2002 Wiley Periodicals, Inc. Biopolymers 64: 236–245, 2002
INTRODUCTION
Cα-Tetrasubstituted α-amino acids are well known as the most effective peptide 310/ α-helix formers and stabilizers.1-7 To this aim even Nature takes advantage of some of them (Aib, α-aminoisobutyric acid or Cα,α-dimethylglycine; Iva, isovaline or Cα-ethyl-Cα-methylglycine) in the family of membrane-active peptaibols (for terminology of peptaibols, see Ref. 8; for review articles, Refs. 9-11; for x-ray diffraction analyses of alamethicin, zervamicins, antiamoebins, and trichogin, Refs. 12, 13-16, 17 and 18, and 19, respectively). The Thorpe–Ingold effect20, 21 has been invoked to explain this structural tendency.22 The question of the preferred helix type (310- vs α-helix) of peptides rich in Cα-tetrasubstituted α-amino acids has been experimentally addressed considering such factors as peptide main-chain length, percentages of Cα-tetrasubstituted α-amino acids, sequence dependence, solvent polarity, temperature, and intermolecular interactions.3-6, 23-27
This work is part of a program aiming at a deeper understanding of the factors mentioned above. In particular, in no peptide system based exclusively on Cα-tetrasubstituted α-amino acids explored so far (with one exception) has the α-helix ever been found, neither in the crystal state nor in solution [the single exception is given by Nα-acylated homo-octapeptide esters based on Cα-methyl-L-valine, L-(αMe)Val, which slowly undergo a 310-helix to α-helix conformational switch in solvents of high polarity under appropriate temperature and concentration conditions28, 29]. In this connection we decided to better explore the effect of the number and positioning of Cα-trisubstituted α-amino acid residues needed to convert a 310-helix into an α-helix in a peptide of a given length. The peptide substrate for this study was chosen to be an Nα-acylated heptapeptide alkylamide (equivalent to an Nα-acylated octapeptide ester in terms of intramolecular H-bond potential) because peptides having this main-chain length are known to be at the interface between 310- and α-helices, and are presumably the most susceptible to this type of helix → helix conformational transformation.3 The only published work on the preferred conformation of Nα-acylated octapeptide esters (or heptapeptide alkylamides) containing a single Cα-trisubstituted α-amino acid deals with a host –(Aib)8– chain (1, Figure 1) with an Aib → L-Leu guest replacement at position 6 (2) (i.e., in an internal position, near the C-terminus of the chain). Results of x-ray diffraction and spectroscopic analyses clearly indicated that the –C(O)–(Aib)5–L-Leu–Aib– sequence 2 is folded in a regular 310-helical structure both in the crystal state and in solution,30-35 as it is the –(Aib)8– parent sequence.36, 37

Chemical formulae of the peptide sequences discussed in this work.
More recently, we have expanded our investigation to the analysis of the effect of a single, internal Cα-trisubstituted α-amino acid positioned near the N-terminus of the peptide chain. To this purpose, a standard, fully Cα-methylated, Nα-acylated heptapeptide alkylamide sequence –C(O)–L-Iva–L-(αMe)Val–L-Iva–L-(αMe)Val–L-(αMe)Phe–L-(αMe)Val–L- Iva–NH– (3) [L-(αMe)Phe, Cα-methyl phenylalanine] was first synthesized and conformationally characterized.38 Both x-ray diffraction and bidimensional NMR/molecular dynamics simulation analyses clearly showed that the peptide adopts a fully developed, regular right-handed 310-helical structure under all experimental conditions tested. In the next step, for an NMR investigation purpose, we designed two singly modified peptide sequences (4 and 5) in which (a) L-(αMe)Val2 was replaced by the Cα-trisubstituted protein L-Val residue (peptide 4), and (b) an L-Abu (Abu, α-aminobutyric acid) residue was substituted for the original L-Iva3 (peptide 5). It is worth noting that in both modified peptides the longest (isopropyl and ethyl, respectively) side chain at the replacement site was preserved. The L-Val2 sequence 4, more soluble in organic solvents, studied by bidimensional NMR/molecular dynamics simulations, was shown to exist in a right-handed 310/α-helical equilibrium with a slight preference for the α-helical conformation.39 Thus, the missing Cα-methylation of Val2 seems to have introduced some flexibility, which had an impact on the overall structure. Conversely, the sparingly soluble mBrBz–L-Iva–L-(αMe)Val–L-Abu–L-(αMe)Val–L-(αMe)Phe–L-(αMe)Val–L-Iva–NHMe 5 (mBrBz, meta-bromobenzoyl; NHMe, methylamino), nicely crystallized out of an acetone solution. Here, we describe details of the x-ray diffraction structure of this L-Abu3 Nα-acylated heptapeptide alkylamide analog. The mBrBz group was incorporated at the N-terminus to help solve the phase problem in the roentgenographic investigation by virtue of its heavy atom (Br). The results are analyzed in parallel with those of the C-terminal sequence Z–L-(αMe)Val–L-(αMe)Phe–L-(αMe)Val–L-Iva–NHMe (6) (Z, benzyloxycarbonyl), the x-ray diffraction structure of which is also described in the present article. This study allowed us to compare the crystal-state conformational preference of this Nα-acylated tetrapeptide alkylamide in isolation with that when it is inserted into a longer peptide sequence.
MATERIALS AND METHODS
Synthesis and Characterization of Peptides
Melting points were determined using a Leitz (Wetzlar, Germany) model Laborlux 12 apparatus and are not corrected. Optical rotations were measured using a Perkin-Elmer (Norwalk, CT) model 241 polarimeter equipped with a Haake (Karlsruhe, Germany) model D thermostat. Thin-layer chromatography was performed on Merck (Darmstadt, Germany) Kieselgel 60F254 precoated plates with the following solvent systems: (1) chloroform/ethanol, 9:1; (2) 1-butanol/acetic acid/water, 3:1:1; (3) toluene/ethanol, 7:1. The chromatograms were examined using ultraviolet fluorescence or developed by chlorine/starch/potassium iodide or ninhydrin chromatic reaction as appropriate. All compounds were obtained in a chromatographically homogeneous state. Their analytical data and physical properties are listed in Table I. All of the synthetic intermediates and the final compound were also characterized by 1H-NMR (data not reported). The chemical structure of the final compound was confirmed by amino acid analysis and matrix-assisted laser desorption/ionization (MALDI) mass spectrometry (data not reported).
| Compound | Yield (%) | Melting Point (°C) | Crystalliz. Solventa | [α]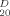 (deg)b (deg)b |
Thin Layer Chromatography | IR (cm−1)c | ||
|---|---|---|---|---|---|---|---|---|
| RF1 | RF2 | RF3 | ||||||
| Z–L-Abu–OH | 55 | 66–68 | DE/PE | −13.4 | 0.45 | 0.90 | 0.25 | 3418, 1743, 1727, 1662, 1546 |
| Z–L-Abu–L-(αMe)Val–L-(αMe)Phe–L-(αMe)Val–L-Iva–NHMe | 34 | 204–205 | EtOAc/PE | −3.8d | 0.70 | 0.95 | 0.35 | 3330, 1697, 1657, 1524 |
| Z–L-(αMe)Val–L-Abu–L-(αMe)Val–L-(αMe)Phe–L-(αMe)Val–L-Iva–NHMe | 48 | 196–198 | CHCl3/PE | −6.4 | 0.70 | 0.90 | 0.30 | 3327, 1702, 1659, 1524 |
| Z–L-Iva–L-(αMe)Val–L-Abu–L-(αMe)Val–L-(αMe)Phe–L-(αMe)Val–L-Iva–NHMe | 66 | 227–228 | EtOAc/PE | 15.4 | 0.65 | 0.90 | 0.30 | 3322, 1696, 1656, 1526 |
| mBrBz–L-Iva–L-(αMe)Val–L-Abu–L-(αMe)Val–L-(αMe)Phe–L-(αMe)Val–L-Iva–NHMe | 71 | 311–312 | CH2Cl2/PE | 14.0e | 0.60 | 0.95 | 0.20 | 3362, 1660, 1522 |
- a DE: diethyl ether; PE: petroleum ether; EtOAc: ethyl acetate.
- b c = 0.5, MeOH.
- c The ir absorption spectra were obtained in KBr pellets; only bands within the 3500–3200 and 1800–1500 cm−1 regions are reported.
- d
[α]
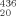 .
.
- e c = 0.2, 2,2,2-trifluoroethanol.
For the large-scale production of the optically pure L-Iva, L-(αMe)Val, and L-(αMe)Phe, we exploited an economically attractive, chemoenzymatic synthesis developed by DSM Research a few years ago.40, 41 It involves a combination of organic synthesis for the preparation of the racemic α-amino acids followed by the use of a broadly specific aminopeptidase to achieve optical resolution. The Z Nα-protected, Cα-methylated α-amino acids42-44 were activated by the acid fluoride method.38, 45, 46 The Z group was removed by catalytic hydrogenation in methanol (MeOH) solution. The synthesis of the mBrBz-heptapeptide 5 was achieved by use of mBrBz-OAt38, 47 (OAt, 1-oxy-7-aza-1,2,3-benzotriazole) in a 9:1 CH2Cl2/CH3CN solvent mixture in the presence of N-methylmorpholine.
X-Ray Diffraction
Colorless crystals of the heptapeptide alkylamide 5 and tetrapeptide alkylamide 6 were grown from methanol and from acetone, respectively, by slow evaporation. Data collection was performed by using a Philips PW 1100 four-circle diffractometer. The two structures were solved by direct methods (SHELXS 97 program48). In the heptapeptide alkylamide 5 refinement was carried out by full-matrix least-squares on F2, using all data, with the SHELXL 97 program.49 In the tetrapeptide alkylamide 6 the full-matrix block least-squares procedure was exploited for refinement. All non-H atoms were refined anisotropically. A planarity restraint was applied to all phenyl rings in both peptides. In the tetrapeptide alkylamide 6 restraints were also imposed to the anisotropic displacement parameters of the phenyl rings to approach isotropic behavior. In both peptides H-atoms were calculated at idealized positions and during the refinement they were allowed to ride on their carrying atom, with Uiso set equal to 1.2 (or 1.5 for methyl groups) times the Ueq of the parent atom. The two highest peaks on the final ΔF map of the heptapeptide alkylamide 5 (0.997 and 0.890 e · Å−3) are located near the Br atom.
Details of the crystallographic data and diffraction parameters for the two structures are given in Table II. Further details of the crystal structures, including final atomic parameters for the non-H atoms, have been deposited with and are available on request from the Director of the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ (England), on quoting the full journal citation.
| Parameter | Heptapeptide Amide (5) | Tetrapeptide Amide (6) |
|---|---|---|
| Empirical formula | C50H77BrN8O8 | C36H53N5O6 |
| Formula weight (amu) | 998.1 | 651.8 |
| Temperature (K) | 293(2) | 293(2) |
| Wavelength (λ) | Cu Kα (1.54178 Å) | Cu Kα (1.54178 Å) |
| Crystal system | Orthorhombic | Monoclinic |
| Space group | P212121 | P21 |
| a (Å) | 9.658(2) | 9.707(3) |
| b (Å) | 18.973(3) | 35.641(8) |
| c (Å) | 28.904(4) | 11.859(5) |
| β (deg) | 90.00 | 113.61(3) |
| V (Å3) | 5296(2) | 3759(2) |
| Z (molecules/unit cell) | 4 | 4 |
| Density (calc.) (g/cm3) | 1.252 | 1.152 |
| Absorption coeff. (mm−1) | 1.523 | 0.634 |
| F(000) | 2128 | 1408 |
| Data collection method | θ–2θ | θ–2θ |
| Crystal size (mm) | 0.30 × 0.20 × 0.20 | 0.40 × 0.30 × 0.10 |
| θ range (deg) | 2.79–60.02 | 2.48–60.02 |
| Index ranges | −1 ≤ h ≤ 10; −1 ≤ k ≤ 21; −32 ≤ 1 ≤ 0 | −10 ≤ h ≤ 9;−1 ≤ k ≤ 40; 0 ≤ l ≤ 13 |
| Reflections collected | 5315 | 6208 |
| Reflections unique | 5151 [R(int) = 0.0549] | 5886 [R(int) = 0.0342] |
| Data/restraints/parameters | 5151/18/614 | 5886/151/829 |
| Goodness-of-fit on F2 | 0.948 | 0.868 |
| Final R indices [I>2(I)] | R1 = 0.0564, wR2 = 0.1353 | R1 = 0.0562, wR2 = 0.1295 |
| R indices (all data) | R1 = 0.0806, wR2 = 0.1480 | R1 = 0.1041, wR2 = 0.1431 |
| Δρ (e/Å3) | 0.997/–0.388 | 0.272/–0.198 |
RESULTS AND DISCUSSION
We determined by x-ray diffraction the molecular and crystal structures of the Nα-acylated heptapeptide alkylamide mBrBz–L-Iva–L-(αMe)Val–L-Abu–L-(αMe)Val–L-(αMe)Phe–L-(αMe)Val–L-Iva–NHMe (5) and its C-terminal tetrapeptide alkylamide sequence Z–L- (αMe)Val–L-(αMe)Phe–L-(αMe)Val–L-Iva–NHMe (6). The molecular structures with the atomic numbering schemes are illustrated in Figures 2 and 3, respectively. Selected backbone and side-chain torsion angles50 are given in Table III. In Table IV the intra- and intermolecular H-bond parameters are listed. For comparison the two tables also report the corresponding data for the parent Nα-acylated heptapeptide alkylamide 3.

The x-ray diffraction structure of mBrBz–L-Iva–L-(αMe)Val–L-Abu–L-(αMe)Val–L-(αMe)Phe–L-(αMe)Val–L-Iva–NHMe (5) with numbering of the backbone atoms. Intramolecular H-bonds are represented by dashed lines.

The x-ray diffraction structures of one (molecule A) of the two independent molecules in the asymmetric unit of Z–L-(αMe)Val–L-(αMe)Phe–L-(αMe)Val–L-Iva–NHMe (6) with numbering of the backbone atoms. Intramolecular H-bonds are represented by dashed lines. In order to align the atom numbering of this tetrapeptide amide with the corresponding atom numbering of the heptapeptide amides, its residues 1–4 have been renumbered 4–7.
| Torsion Angle | Heptapeptide Amide (3) | Heptapeptide Amide (5) | Tetrapeptide Amide (6) | ||
|---|---|---|---|---|---|
| Mol. A | Mol. B | Mol. A | Mol. B | ||
| ϕ1 | −61.5 (10) | −55.4 (15) | −52.0 (7) | ||
| ψ1 | −29.9 (10) | −32.1 (13) | −44.5 (7) | ||
| ω1 | −171.4 (7) | −170.8 (10) | −172.7 (5) | ||
| ϕ2 | −52.8 (9) | −57.0 (15) | −53.8 (7) | ||
| ψ2 | −32.3 (8) | −29.6 (13) | −40.7 (7) | ||
| ω2 | −175.3 (6) | −179.5 (9) | −172.5 (5) | ||
| ϕ3 | −51.0 (9) | −48.4 (14) | −67.6 (7) | ||
| ψ3 | −41.6 (9) | −33.1 (12) | −43.5 (7) | ||
| ω3 | −171.2 (7) | −170.9 (8) | −179.6 (5) | ||
| ϕ4 | −54.7 (9) | −54.0 (10) | −57.3 (6) | −55.1 (6) | −57.3 (6) |
| ψ4 | −32.7 (9) | −26.3 (9) | −45.4 (6) | −31.6 (6) | −28.6 (6) |
| ω4 | −175.0 (7) | −176.8 (6) | −175.8 (4) | −172.9 (4) | −174.6 (4) |
| ϕ5 | −53.1 (9) | −51.2 (8) | −58.5 (6) | −58.1 (6) | −53.7 (6) |
| ψ5 | −32.9 (9) | −37.9 (8) | −49.0 (6) | −20.4 (6) | −28.0 (6) |
| ω5 | −178.1 (7) | −178.2 (6) | −173.9 (5) | 174.2 (4) | −179.1 (4) |
| ϕ6 | −51.6 (9) | −51.2 (9) | −53.8 (7) | −47.5 (6) | −49.9 (6) |
| ψ6 | −45.1 (9) | −41.6 (9) | −44.2 (8) | −40.1 (6) | −35.7 (6) |
| ω6 | −175.5 (8) | −176.5 (7) | −174.1 (7) | −175.5 (5) | −178.1 (5) |
| ϕ7 | −78.012) | −56.2 (10) | −51.1 (10) | −67.1 (6) | −63.7 (6) |
| ψ7 | −8.0 (15) | −42.8 (10) | −49.0 (10) | −12.0 (7) | −17.8 (7) |
| ω7 | −178.1 (12) | −176.6 (9) | −175.5 (9) | 178.5 (4) | 179.1 (5) |
| Side chains | |||||
| χ11 | −74.0 (10) | 176.6 (21) | 177.2 (7) | ||
| χ21,1 | 60.9 (9) | 168.1 (16) | −177.0 (7) | ||
| χ21,2 | −171.9 (8) | −75.2 (28); 42.1 (19) | 56.3 (8) | ||
| χ31 | 170.9 (9) | 165.6 (13) | −61.9 (7) | ||
| χ41,1 | 169.2 (9) | 64.7 (9) | −67.6 (6) | 59.5 (6) | 62.7 (6) |
| χ41,2 | −64.0 (9) | −171.2 (9) | 167.8 (5) | −174.8 (5) | −172.1 (5) |
| χ51 | −178.8 (8) | 176.8 (6) | −172.9 (2) | −54.6 (3) | −52.5 (4) |
| χ52,1 | 92.3 (8) | 89.9 (7) | 131.5 (3) | 88.7 (2) | 95.5 (3) |
| χ52,2 | −88.0 (9) | −88.0 (7) | −53.2 (5) | −88.8 (5) | −86.7 (6) |
| χ61,1 | 166.6 (8) | 171.4 (9) | −70.3 (7) | −70.6 (5) | −71.7 (5) |
| χ61,2 | −70.6 (9) | −61.5 (9) | 162.5 (6) | 162.4 (5) | 163.7 (5) |
| χ71 | −61.6 (10) | −178.9 (15); −78.8 (21) | 175.5 (10) | −67.7 (8) | −69.3 (7) |
- a Taken from Ref. 38.
- b In order to align the torsion angles of this tetrapeptide amide with the corresponding torsion angles of the heptapeptide amides, its residues 1–4 have been renumbered 4–7.
| Donor | Acceptor | Length (Å) (N … O) | Length (Å) (H … A) | Angle (°) (N—H … O) | Symmetry Operation | |
|---|---|---|---|---|---|---|
| Heptapeptide amide (3) | ||||||
| Intramolecular | N3A | O0Ab | 3.335 (10) | 2.50 | 165 | x, y, z |
| N4A | O1A | 3.084 (8) | 2.23 | 155 | x, y, z | |
| N5A | O2A | 3.068 (8) | 2.28 | 152 | x, y, z | |
| N6A | O3A | 3.185 (8) | 2.37 | 158 | x, y, z | |
| N7A | O4A | 2.962 (9) | 2.26 | 138 | x, y, z | |
| NTAb | O5A | 2.864 (13) | 2.07 | 152 | x, y, z | |
| N3B | O0Bb | 3.246 (11) | 2.43 | 159 | x, y, z | |
| N4B | O1B | 3.004 (9) | 2.15 | 171 | x, y, z | |
| N5B | O2B | 3.136 (10) | 2.28 | 174 | x, y, z | |
| N6B | O3B | 3.132 (8) | 2.32 | 158 | x, y, z | |
| N7B | O4B | 2.995 (8) | 2.27 | 142 | x, y, z | |
| NTBb | O5B | 2.985 (9) | 2.36 | 130 | x, y, z | |
| Head-to-tail | N1A | O6B | 2.892 (8) | 2.04 | 171 | 1+x, y, 1+z |
| N2A | O7B | 3.232 (9) | 2.61 | 130 | 1+x, y, 1+z | |
| N1B | O7A | 2.949 (11) | 2.33 | 129 | x, y, z | |
| Heptapeptide amide (5) | ||||||
| Intramolecular | N3 | O0b | 2.969 (6) | 2.27 | 139 | x, y, z |
| N4 | O1 | 3.199 (6) | 2.69 | 120 | x, y, z | |
| N5c | O1 | 3.340 (5) | 2.50 | 166 | x, y, z | |
| N6 | O2 | 3.270 (6) | 2.45 | 159 | x, y, z | |
| N7 | O3 | 3.429 (6) | 2.63 | 156 | x, y, z | |
| N7 | O4 | 3.258 (7) | 2.68 | 126 | x, y, z | |
| NTb | O5 | 3.089 (8) | 2.51 | 126 | x, y, z | |
| Head-to-tail | N1 | O6 | 2.892 (6) | 2.06 | 164 | −x−1/2, −y, z−1/2 |
| Tetrapeptide amide (6) | ||||||
| Intramolecular | N6Ad | O3Ab | 3.110 (6) | 2.26 | 173 | x, y, z |
| N7A | O4A | 3.063 (5) | 2.23 | 162 | x, y, z | |
| NTAb | O5A | 2.898 (7) | 2.07 | 161 | x, y, z | |
| N6B | O3Bb | 3.142 (5) | 2.29 | 172 | x, y, z | |
| N7B | O4B | 3.088 (6) | 2.26 | 161 | x, y, z | |
| NTBb | O5B | 2.891 (6) | 2.07 | 160 | x, y, z | |
| Head-to-tail | N4A | O7A | 2.858 (6) | 2.16 | 139 | x, y, z+1 |
| N4B | O7B | 2.891 (5) | 2.20 | 137 | x+1, y, z+1 |
- a Taken from Ref. 38.
- b O0 in the heptapeptide amides (and O3 in the tetrapeptide amide) is the carbonyl oxygen atom preceding the N-terminal residue; in all peptides NT is the amide nitrogen atom following the C-terminal residue.
- c The atoms involved in an intramolecular H-bond forming an α(C13)-turn, instead of a β(C10)-turn, are in bold.
- d In order to align the atom numbering of this tetrapeptide amide with the corresponding atom numbering of the heptapeptide amides, its residues 1–4 have been renumbered 4–7.
The molecules of the Abu3 heptapeptide amide 5 are fully-developed right-handed helices, stabilized by a large set of intramolecular, consecutive, or bifurcated, CO · · · H—N H-bonds. The right-handed helical screw sense is in particular dictated by the known conformational bias of the L-Iva (with a linear side chain)7, 38, 44 and L-(αMe)Val (with a β-branched side chain)7, 38, 46 residues. The average values for the seven sets of backbone ϕ,ψ torsion angles are −56.3°, −45.2°. The average ϕ value is closer to that typical of a 310-helix (−57°) than to that of an α-helix (−63°).23 However, the opposite holds true for the average ψ value (−30° for a typical 310-helix and −42° for a typical α-helix). This mixed 310/α-helical structure, suggested by the backbone torsion angles, is confirmed by the analysis of the intramolecular H-bonds, although some of them are weak,51-53 particularly if part of the bifurcation sites (in any case, intramolecular H-bonds with an N · · · O distance > 3.0 Å are a common observation in 310- and α-helices). At the N-terminus the molecular conformation displays a short 310-helix with two consecutive type III β-turn (C10) structures.54-56 The CO group of the mBrBz group forms a H-bond with the L-Abu3 N–H group, while the L-Iva1 CO group is involved in a H-bond with the L-(αMe)Val4 N—H group. However, the L-Iva1 CO group is also involved in a weak interaction with the L-(αMe)Phe5 N—H group giving rise to an α-turn (C13) conformation.57-59 Then, this region of bifurcation is followed by two weakly stabilized, consecutive α-turn structures involving carbonyl oxygens of L-(αMe)Val2 and L-Abu3 and amino groups of L-(αMe)Val6 and L-Iva7. A second site of bifurcated arrangement is seen at the C-terminus, as the L-Iva7 amino group is also involved in the formation of a weakly stabilized β-turn with the L-(αMe)Val4 CO group. The short C-terminal 310-helical segment ends up with a H-bond between the methylamido N—H and the L-(αMe)Phe5 carbonyl groups.
In the tetrapeptide amide 6 each independent molecule A and B is folded in a right-handed 310-helix stabilized by three intramolecular CO · · · H—N H-bonds. Two regular type III β-turns are followed by a C-terminal, distorted β-turn in which the values of the ϕ,ψ torsion angles of the L-(αMe)Val and L-Iva residues are intermediate between those of a type I and a type III β-turn (position i+2). The conformational differences, including side-chain dispositions (χ torsion angles),60 between molecules A and B of the tetrapeptide amide are only of minor significance.
For reason of comparison, in Tables III and IV the backbone and side-chain torsion angles, and the intra- and intermolecular H-bond parameters, respectively, for the regular 310-helical, parent L-Iva3 heptapeptide amide (3) (independent molecules A and B)38 are also listed. Significant backbone conformational differences among the five molecules are observed: (a) in the ψ4 and ψ5 torsion angles (the absolute values of which are consistently higher in the L-Abu3 heptapeptide amide 5) and (b) in the related, α-turn type, O2 · · · H6—N6 and O3 · · · H7—N7 intramolecular H-bonds (seen only in the L-Abu3 heptapeptide amide 5) and β-turn type O3 · · · H6—N6 intramolecular H-bond (seen only in the L-Iva3 heptapeptide amide 3 and in the tetrapeptide amide 6). From this comparison, it may be concluded that this Nα-acylated tetrapeptide amide segment may adopt either a mixed α/310-helical structure (when incorporated at the C-terminus of the L-Abu3 heptapeptide amide 5) or an essentially 310-helical structure (either when in isolation or when incorporated at the C-terminus of the fully Cα-methylated L-Iva3 heptapeptide amide 3).
The packing mode of the heptapeptide amide 5 is characterized by an intermolecular H-bond between the N1 group and the O6 carbonyl oxygen atom of a (−x − ½, −y, z− ½) symmetry related molecule. Rows of molecules, head-to-tail H-bonded and related through a crystallographic twofold screw axis, are thus formed through the c direction.
In the packing mode of the tetrapeptide amide 6 head-to-tail intermolecular H-bonds are observed between molecules of the same type. More specifically, the N4A—H group is H-bonded to the O7A carbonyl oxygen atom of a (x + 1, y, z + 1) translational equivalent of molecule A, while the N4B—H group is H-bonded to the O7B carbonyl oxygen atom of a (x + 1, y, −z + 1) translational equivalent of molecule B. As a consequence, rows of molecules A are formed along the c direction, while rows of molecules B are observed along the ac direction. Rows of the two kinds alternate along the b direction.
CONCLUSIONS
This paper clearly shows that in the crystal state an Nα-acylated heptapeptide alkylamide, containing six Cα-tetrasubstituted α-amino acids and a single Cα-trisubstituted residue near the N-terminus (L-Abu3) (5), tends to fold in a well-developed α-helical structure in the central region, although this segment is preceded and followed by two short 310-helical segments. This result is in good agreement with the published NMR data on the L-Val2 analog 439 of the same, fully 310-helical, parent peptide 3,38 and beautifully parallels that of an NMR study on an Ala-rich peptide, where an α-helical sequence was identified in the middle and short 310-helical stretches at both termini.61
A previous detailed structural investigation on an Nα-acylated octapeptide ester (equivalent to a heptapeptide alkylamide as far as its capability of intramolecular H-bond formation is concerned), characterized by seven Aib residues and a single Cα-trisubstituted α-amino acid (L-Leu6) near the C-terminus (2), unequivocally established the onset of a fully developed, rigid, regular 310-helical conformation,30-35 as observed in the –(Aib)8– homopeptide sequence (1).36, 37 Taken together, these findings support the view that, even in the case of a very high percentage of Cα-tetrasubstituted α-amino acids discussed here, the precise positioning of the single Cα-trisubstituted residue in the sequence may have a dramatic effect on the helical structure of the peptide backbone. More specifically, incorporation of the guest residue near the N-terminus seems critical for the observation of the conformational transition. Even if in a single, specific case (an Nα-tert-butyloxycarbonylated or Nα-acetylated heptapeptide methyl ester characterized by as many as six Cα-trisubstituted amino acids) it has been reported that in the crystal state the nature of the Nα-blocking may play a role on the type of helical structure adopted,62 it is evident that in our heptapeptide methylamides 3–5 such an effect cannot be operative as all three compounds are Nα-acylated with the same moiety (mBrBz).
Other interesting information extracted from this work is that the preferred type of helical structure of even a fully Cα-tetrasubstituted, Nα-acylated tetrapeptide alkylamide, as 6, may be governed by the presence/absence of additional residues in the sequence.

