

Versatile Halide-Pair-Driven Multicomponent Polymerization for Library Synthesis of Sequence-Controlled Semiconducting Dendronized Polymers
Graphical Abstract
A halide-pair-driven multicomponent polymerization (MCP) strategy enables the efficient synthesis of diverse sequence-controlled semiconducting poly(triarylamine)s (PTAAs), including dendronized variants, advancing the development of next-generation organic materials.

Abstract
Sequence-controlled semiconducting polymers represent a new frontier in organic electronics, where precise molecular sequence directly dictates device performance. However, achieving both high sequence fidelity and structural diversity remains a significant challenge using conventional synthetic protocols. To address this issue, we introduce a versatile halide-pair-driven multicomponent polymerization (MCP) strategy that enables the library synthesis of sequence-controlled semiconducting poly(triarylamine)s (PTAAs). By optimizing halide pairing in conjunction with a rationally designed Buchwald ligand–Pd system featuring catalyst-transfer capability, we achieved efficient sequential cascade aminations, thereby enabling the MCP. The versatility of this strategy was demonstrated through the synthesis of a library of sequence-controlled PTAAs, including dendronized variants, underscoring its potential as a general platform for functional semiconducting material discovery.
Introduction
Analogous to the specific sequence of nucleotides in DNA that encodes genetic information, the sequence of synthetic polymers also plays a pivotal role in defining their properties and functions.[1-3] Various approaches have been developed to regulate polymer sequences, including template-assisted synthesis, iterative coupling, and reactivity-controlled step-growth polymerizations. Notably, Ueda and coworkers demonstrated that structural regularity of polymers such as polyamides can be finely tuned even in one-step condensation polymerizations by exploiting the inherent reactivity differences between monomers, thereby providing an elegant route to sequence-controlled polymerization.[4-6] Building on these concepts, recent efforts have focused on achieving precise sequence control in semiconducting polymers, where even subtle variations in monomer arrangement can significantly influence optoelectronic properties such as bandgap and charge carrier mobility.[7-10] Although alternating architectures are often accessible through step-growth polymerizations of two distinct monomers, the development of versatile synthetic strategies capable of constructing semiconducting polymers with three or more distinct monomers arranged in a precisely defined sequence remains a major synthetic challenge.
Poly(triarylamine) (PTAA) hold significant prominence in various state-of-the-art applications such as organic light-emitting diodes, perovskite solar cells, electrochromic devices, stretchable electronics, and battery electrodes.[11-18] Accordingly, the development of diverse PTAA materials with precisely controlled microstructures has been actively pursued to enhance their performance.[19-24] However, existing synthetic methods that allow for both diversity and precision often require time-consuming, multi-step processes. For example, the widely used Buchwald–Hartwig amination polymerization necessitates a stepwise approach to construct sequence-controlled PTAAs. In particular, isolating the bis-amine intermediate in high purity remains a challenging step, necessitating tedious purification procedures and thereby increasing production costs. Therefore, a versatile one-shot polymerization strategy employing readily available starting materials is highly desirable to address these challenges.
Multicomponent polymerization (MCP) is a powerful synthetic method that combines three or more monomers in a cascade manner within a single flask, enabling the rapid generation of diverse and complex polymer architectures (Scheme 1a).[25-29] Over the past decade, MCP has emerged as a key strategy in diversity-oriented polymerization, enabling the construction of polymer libraries applicable to photopatterning, chemical sensing, and bioimaging.[30-32] Nevertheless, developing a general MCP strategy for the library synthesis of widely used semiconducting polymers, including PTAAs, remains a major challenge. In addition, introducing complex microstructures such as dendronized semiconducting polymers via diversity-oriented MCP has been a great challenge, with only one successful example reported to date, which yielded a non-conjugated backbone.[33]

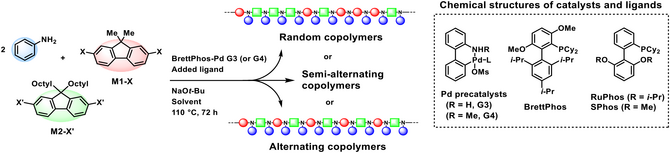
To realize the MCP for various alternating PTAAs, we designed the initial reaction mixture consisting of three components: arylamine and two different aryl dihalides (X-Ar1-X and X’-Ar2-X’). In this case, the selective and rapid in situ formation of a secondary amine intermediate (HRN-Ar1-NRH) from X-Ar1-X and R-NH2, without the involvement of the spectator monomer, X’-Ar2-X’, is crucial. This is because the purity of the intermediate plays a pivotal role in maintaining the stoichiometric balance and minimizing side reactions during step-growth polymerization, ultimately enabling the formation of high molecular weight polymers with minimal sequence defects.[34] To this end, we devised three strategies for the in situ generation of the key intermediate, HRN-Ar1-NRH, in high purity. First, there must be a strong kinetic preference for the coupling reaction between the arylamine and one of the aryl dihalides, specifically X-Ar1-X, over the other aryl dihalide, X’-Ar2-X’. This can be accomplished by selecting an appropriate combination of X and X’ halides (Schemes 1b and 1d).[35, 36] Second, the intramolecular oxidative addition, or catalyst-transfer reaction, can facilitate the conversion of the mono-functionalized intermediate into the bis-functionalized product (Schemes 1c and 1d).[37-43] This process requires the careful selection of ligands that support not only efficient catalyst-transfer but also effective amination with both primary and secondary amines. Third, the use of a Buchwald precatalyst ensures the rapid generation of highly active Pd(0)–ligand species with excellent purity, thereby enabling the efficient in situ formation of the key intermediate in high yield.[44-47]
Herein, we present a rationally designed MCP strategy for the library synthesis of sequence-controlled semiconducting PTAAs. Through mechanistic studies, we elucidated the individual roles of halide pairing, catalyst-transfer, and precatalyst selection in achieving high molecular weight polymers with well-defined sequences. Moreover, this strategy accommodates a wide variety of monomers, thereby underscoring its versatility as a platform for constructing sequence-controlled semiconducting PTAA libraries. Notably, this strategy enables access not only to singly dendronized, but even to doubly dendronized semiconducting PTAAs with a high degree of structural complexity but with well-defined alternating backbones, which are rarely accessible through conventional synthetic methods (Scheme 1e).
Results and Discussion
To realize and optimize the MCP, the model polymerization was designed using two equivalents of aniline, one equivalent of 2,7-dihalide-9,9-dimethylfluorene (M1-X where X═Br or I), and one equivalent of 2,7-dihalide-9,9-dioctylfluorene (M2-X’ where X’═Br or Cl) (Table 1). The mixed ligand catalytic system consisting of the BrettPhos Pd G3 precatalyst and the additional RuPhos ligand was used to facilitate the coupling reaction of both primary and the in situ generated secondary amines.[36] Under these conditions, we systematically varied the halide combinations of the two monomers to evaluate their effects on polymerization efficiency and selectivity. As a first case, polymerization using M1-Br and M2-Br afforded a polymer with a weight average molecular weight (Mw) of 44.2 kDa, demonstrating efficient polymerization under the applied catalytic system (Table 1, entry 1). When the monomer combination was changed to M1-I and M2-Br, a polymer with a higher Mw of 106.3 kDa was obtained (Table 1, entry 2). The polymerization using either M1-Br or M1-I in combination with M2-Cl produced polymers with relatively lower Mw values of 13.6 and 14.8 kDa, respectively (Table 1, entries 3 and 4).
 |
|||||||||
| Halide | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | X | X’ | Precatalyst (2 mol%)a) | Ligand (mol%)a) | Solvent (v/v) | Mn (kDa)b) | Mw (kDa)b) | Ðb) | %Alternationc) |
| 1 | Br | Br | BrettPhos G3 | RuPhos (2) | 1,4-dioxane | 15.3 | 44.2 | 1.68 | 54 |
| 2 | I | Br | BrettPhos G3 | RuPhos (2) | 1,4-dioxane | 46.8 | 106.3 | 2.31 | 53 |
| 3 | Br | Cl | BrettPhos G3 | RuPhos (2) | 1,4-dioxane | 6.8 | 13.6 | 2.04 | 73 |
| 4 | I | Cl | BrettPhos G3 | RuPhos (2) | 1,4-dioxane | 7.5 | 14.8 | 1.98 | 89 |
| 5 | I | Cl | BrettPhos G4 | RuPhos (2) | 1,4-dioxane | 9.2 | 18.3 | 2.00 | n.d. |
| 6 | I | Cl | BrettPhos G4 | RuPhos (2) | toluene/t-AmOH (5/1) | 9.5 | 22.8 | 2.39 | n.d. |
| 7 | I | Cl | BrettPhos G4 | SPhos (2.4) | toluene/t-AmOH (5/1) | 10.3 | 24.3 | 2.37 | 91 |
| 8d) | I | Cl | BrettPhos G4 | SPhos (2.4) | toluene/t-AmOH (5/1) | 11.4 | 25.6 | 2.25 | 91 |
- a) The concentrations of precatalysts and ligands are determined based on the concentration of aniline.
- b) Determined by tetrahydrofuran (THF) size exclusion chromatography (SEC) calibrated using polystyrene (PS) standards.
- c) Determined by 13C NMR spectroscopy (%Alternation = ).
- d) Precipitated into acetone. Other samples in entries 1−7 are precipitated into MeOH.
Subsequently, the microstructures of the obtained polymers were analyzed by 13C NMR spectroscopy (Figure 1). As the reference, the alternating copolymer, P1, with a Mw of 31.5 kDa was prepared using the conventional two-step protocol (Figure 1a). The 13C NMR spectrum showed the discrete number of signals corresponding to the structures of the alternating copolymer. In contrast, the 13C NMR spectrum of the polymer obtained from the one-shot polymerization of M1-Br and M2-Br exhibited a greater number of aromatic signals—particularly at 152.0, 148.3, 146.6, 136.2, and 133.8 ppm—which may indicate a random copolymerization of M1-Br and M2-Br (Figure 1b). Based on the 13C NMR analysis, the calculated % alternation was 54%, consistent with a statistical copolymerization (Table 1, entry 1). This value was quantified by analyzing the relative integral values of diagnostic signals corresponding to alternating and homodimeric structures as detailed in the Supporting Information (SI page S8). A similar random copolymerization behavior was observed for the combination of M1-I and M2-Br (Figure 1c). However, when M1-Br and M2-Cl were used for polymerization, the number of signals observed in the 13C NMR spectrum was comparable to that of the alternatingly sequenced polymer P1 (Figure 1d). Nevertheless, a closer inspection of the 13C NMR spectrum of the polymer derived from M1-Br and M2-Cl revealed small shoulder peaks adjacent to the main signals, indicative of minor sequence irregularities, with the % alternation reaching only 73%. In contrast, the one-shot polymerization of M1-I and M2-Cl yielded a polymer with a % alternation of 89, whose 13C NMR spectrum closely matched that of the well-defined alternating copolymer P1 synthesized via the conventional two-step protocol (Figure 1e). This enhanced sequence selectivity may be attributed to the greater disparity in halide reactivity between M1-I and M2-Cl, which is the largest among the tested halide pairs.

Although the monomer combination of M1-I and M2-Cl produced the well-defined alternating copolymer P1, its Mw (14.8 kDa) was rather lower than that of P1 (31.5 kDa) synthesized via the conventional two-step protocol. Therefore, further optimization was undertaken to retain the alternating sequence while increasing the molecular weight of P1. First, replacing the G3 precatalyst with the BrettPhos G4 precatalyst increased the Mw from 14.8 to 18.3 kDa (Table 1, entry 5). This improvement is likely due to differences in the activation mechanism: the G3 precatalyst generates BrettPhos–Pd(0) and free carbazole upon activation, with carbazole potentially acting as a terminator by competing with the in situ–generated secondary amine intermediate in the step-growth process (Figure 2a).[44, 45] In contrast, the G4 precatalyst releases N-methylcarbazole upon activation, thus eliminating termination via free carbazole. To further support this idea, polymerization was conducted using the G4 precatalyst with an externally added 1–5 mol% of carbazole relative to the amount of aniline monomer (Figure 2b). Indeed, as the amount of carbazole increased in the reaction mixture, the Mw of P1 gradually decreased to less than 1 kDa, further supporting the superior role of the G4 precatalyst over the G3 precatalyst in polymerization. Additionally, replacing 1,4-dioxane with a co-solvent of toluene and tert-AmOH increased the Mw from 18.3 to 22.8 kDa (Table 1, entry 6). Furthermore, substituting the added ligand from RuPhos (2.0 mol%) to SPhos (2.4 mol%) further increased the Mw to 24.3 kDa while maintaining a high %alternation of 91 (Table 1, entry 7 and Figure 1f). Finally, fractional precipitation of P1 into acetone, instead of methanol, effectively removed low molecular weight oligomers without significantly compromising the overall yield, leading to a final Mw of 25.6 kDa (Table 1, entry 8 and Figure S1). Based on the relative number average molecular weight (Mn) value obtained by SEC (calibrated with PS standards) and a repeat unit molecular weight of 763.12 g mol−1, the degree of polymerization (DP) of P1 was approximately estimated to be 15 (Table 1, entry 8).

To achieve MCP, we assumed two key strategies: exploiting the difference in halide reactivity and utilizing the catalyst-transfer process. To evaluate the effectiveness of these strategies within the polymerization mechanism, we initially conducted a kinetic study on the three-component reaction involving p-toluidine (2.2 equiv.), p-iodotoluene (1.0 equiv.), and p-bromotoluene (1.0 equiv.) (Figure 3a). Kinetic analysis using 1H NMR revealed that both halide monomers reacted simultaneously, although 4-iodotoluene was consumed faster than p-bromotoluene (Figure 3a). In contrast, when the halide combination was changed from I/Br to Br/Cl, the consumption rates of p-bromotoluene and p-chlorotoluene were vastly different, with less than 10% conversion of p-chlorotoluene until p-bromotoluene was fully consumed (Figure 3a). Moreover, when the halide combination was altered from Br/Cl to I/Cl, the difference became even larger, showing less than 2% conversion of p-chlorotoluene until p-iodotoluene was fully consumed (Figure 3a). In addition, detailed ¹H NMR analysis of the I/Cl system confirmed that tertiary amine formation was negligible, further supporting the selective formation of secondary amine intermediates that underpins sequence control in the MCP process (SI page S12). These observations indicate that the choice of halide combinations can regulate the sequence selectivity within the polymer, with the I/Cl combination demonstrating the highest purity of the alternating sequence.

Next, to understand the role of the catalyst-transfer process in the MCP, small molecule amination reactions were conducted using p-toluidine and three different p-dihalobenzenes (X═I, Br, and Cl), with kinetic monitoring of the reactions (Figure 3b). When p-diiodobenzene was used, the bis-functionalized product was exclusively formed at all time points, indicating that the intramolecular catalyst-transfer process dominated throughout the reaction. Similarly, with p-dibromobenzene, the bis-functionalized product remained the major product, although a slightly higher amount of the mono-functionalized intermediate was detected compared to the p-diiodobenzene reaction (Figure 3b). In contrast, when p-dichlorobenzene was employed, the mono-functionalized intermediate was initially formed and subsequently converted to the bis-functionalized product (Figure 3b). This suggests that the catalyst-transfer process is less favorable with p-dichlorobenzene, while also highlighting its higher reactivity compared to the mono-functionalized product. Consequently, p-diiodobenzene most effectively promotes the catalyst-transfer process, leading to the highest ratio of bis- to mono-functionalized intermediates.
Building on this mechanistic framework, we conducted time-dependent 1H NMR analysis of the model MCP. This study revealed the selective formation of a bis-functionalized intermediate (90% NMR yield at 6 min), accompanied by complete consumption of p-diiodobenzene and approximately 95% conversion of p-toluidine, without any observable consumption of M2-Cl (Figure 3c). Between 6 and 7 min, a minor reaction involving the residual p-toluidine and M2-Cl occurred after the initial consumption phase, accounting for the slight decrease in alternating sequence selectivity. This may be attributed to a potential stoichiometric imbalance despite careful weighing. Subsequently, the concurrent consumption of the bis-functionalized intermediate and M2-Cl was observed, confirming that the progression of step-growth polymerization.
Although a catalyst-transfer process was observed during the developed MCP, its specific contribution to sequence control remains uncertain. Accordingly, to determine whether the high degree of sequence alternation originates solely from the intrinsic reactivity difference between iodide and chloride, or from a synergistic effect between halide pairing and the catalyst-transfer process, an MCP was conducted using a mono-functionalized intermediate, aniline, and M2-Cl (Figure 4). In the case where the intermediate preferentially undergoes amination with aniline over M2-Cl, an alternating copolymer would be produced. Conversely, competitive amination involving both the intermediate and M2-Cl with aniline would compromise sequence selectivity. Indeed, 13C NMR analysis of the resulting polymer revealed a high degree of sequence alternation, comparable to that obtained from the MCP using the diiodo compound (Figure 4). This finding suggests that, although the catalyst-transfer process can facilitate the formation of bis-functionalized intermediates during MCP, its influence on sequence selectivity is secondary. Instead, the intrinsic reactivity difference between iodide and chloride serves as the primary determining factor.

Based on mechanistic insights, MCP is proposed to proceed through a series of chemoselective amination reactions (Figure 5a). Initially, the primary amine preferentially undergoes amination with the aryl diiodide over the aryl dichloride, as oxidative addition to the aryl diiodide occurs at a significantly faster rate. Following this, the Pd(0)–L catalyst remains coordinated to the mono-functionalized intermediate via π-complex formation, facilitating intramolecular oxidative addition (i.e., catalyst-transfer) and enabling the highly selective formation of the bis-functionalized intermediate. The bis-functionalized intermediates then undergo step-growth polymerization with aryl dichlorides, yielding alternating PTAAs. In contrast, when two aryl dibromides are employed, the intermolecular oxidative addition proceeds without chemoselectivity from the outset (Figure 5b). Consequently, despite the occurrence of the catalyst-transfer process, a variety of intermediates are formed, resulting in random copolymerization. These findings highlight the critical role of halide pairing in achieving precise sequence control.

After identifying key factors contributing to the effectiveness of the MCP strategy, we explored diversity-oriented polymerization to construct a library of sequence-controlled PTAAs using readily available starting materials (Figure 6). To investigate the electronic effects of substituents on polymerization, we employed various para-substituted anilines with electron-donating (-butyl, -OMe) and electron-withdrawing (-F) groups. These polymerizations yielded polymers P2 − P4 with Mw ranging from 12.9 to 16.8 kDa and yields of 73%−85%. Moreover, polymerization with n-hexyl amine proceeded smoothly, yielding polymer P5 with an Mw of 17.4 kDa. After confirming the robustness of the MCP method for a broad scope of amines, we then substituted 9,9-dimethyl-2,7-diiodo-fluorene (B1) with (bi)phenyl diiodides and performed MCP. This modification was motivated by the structural similarity of alternating copolymers containing (bi)phenyl and fluorene units to PF8-TAA, a well-known hole-transporting material used in optoelectronic applications such as perovskite solar cells and organic light-emitting diodes.[48-50] Specifically, the combinations of aniline derivatives (A1, A3, and A4), 4,4′-diiodobiphenyl (B3), and 9,9-dioctyl-2,7-dichlorofluorene (C1) resulted in polymers P6 − P8 with Mw ranging from 14.9 to 32.8 kDa. Similarly, polymerization with 1,4-diiodobenzene (B4) instead of B3 proceeded smoothly regardless of the type of amine, yielding polymers P9−P13 with Mw ranging from 7.4 to 28.2 kDa. Additionally, the opposite combination of halide units, 9,9-dimethyl-2,7-diiodo-fluorene (B1) and 4,4′-dichlorobiphenyl (C2), was also effective in producing polymers with Mw of 21.2 kDa (P14) and 18.4 kDa (P15). Thereafter, aromatic units containing heteroatoms were introduced into the MCP to further examine the versatility of the developed method. The polymerizations of various amines (A1–A5), N-containing carbazole (B5), and C1 were performed, which resulted in polymers P16−P20 with Mw ranging from 11.5 to 26.2 kDa. Furthermore, polymerizations with the electron-deficient monomer, 4,4′-dichlorodiphenyl sulfone (C3), also resulted in the unique donor–acceptor type polymers P21−P24 with Mw ranging from 6.2 to 20.5 kDa.

The sequence of the synthesized polymer library was examined using 13C NMR spectroscopy. As a reference, alternating copolymers with representative repeating units were selected and synthesized using the conventional two-step protocol (5 different polymers: P1, P6, P7, P9, and P16; for details, see Table S1 and SI pages S54, S60, S61, S64, and S72). In summary, this analysis confirmed that the 13C NMR spectra of the polymers synthesized via both the MCP and the conventional two-step method were in close agreement, demonstrating the versatility of MCP in producing well-defined alternating PTAAs.
The fundamental properties of the synthesized PTAAs were investigated. DSC analyses revealed that the PTAAs are amorphous, exhibiting glass transition temperatures (Tg) ranging from 66 to 154 °C (Figure 6). The Tg of PTAAs was primarily governed by the rigidity of the polymer backbone (Tables S2 and S3). Among the tested structures, incorporation of N-ethylcarbazole led to the highest Tg, followed by biphenyl and dimethylfluorene units, with phenyl yielding the lowest values. This trend was consistently observed regardless of the para-substituent on the phenyl side chains (R═H, n-Bu, OMe, and F). In contrast, PTAAs incorporating aliphatic amines, such as n-hexylamine, exhibited significantly lower Tg values, which is attributed to the enhanced segmental mobility imparted by the flexible hexyl chains. The UV–vis analysis of the PTAAs in film state showed λmax values ranging from 382 to 414 nm, with typical optical bandgaps (Eg) of approximately 2.9 eV (SI page S28). The highest occupied molecular orbital (HOMO) levels, determined by cyclic voltammetry, ranged from − 5.3 to − 4.8 eV (Figure 6). The P21−P24 prepared with the electron-withdrawing unit (C3) exhibited relatively deeper HOMO levels from −5.3 to −5.1 eV. Moreover, the P21−P24, consisting of the donor–acceptor type repeat units, exhibited fluorescence emission in chloroform, with an emission maximum around 500 nm upon excitation at 370 nm (SI page S42).
Dendrimers are well-defined, monodisperse macromolecules characterized by extensive branching and a high density of functional groups.[51-53] When their subunits, dendrons, are grafted onto a linear polymer backbone, the resulting dendronized polymers form molecular architectures that bridge the gap between linear polymers and colloidal particles.[54-61] As a result, these materials exhibit unique conformational and aggregation behaviors, which can be systematically tuned by varying the size, type, and placement of dendrons. Notably, dendronized semiconducting polymers are regarded as molecular nanowires, integrating the unique properties of both the conjugated backbone and the peripheral dendron units.[62-66] Nevertheless, despite their promising potential, synthetic strategies employing MCP for the precise and structurally diverse construction of dendronized semiconducting polymers remain scarce. To address this issue, we aimed to synthesize dendronized PTAAs incorporating Fréchet-type G1−G3 benzyl ether dendron units via the developed MCP method (Figure 7a). Specifically, we designed dendronized PTAAs containing p-toluidine, carbazole, and fluorene units in the backbone. As exemplified by the successful synthesis of the G3 dendron, the resulting polymer exhibited Mw ranging from 12.0 to 22.1 kDa despite the significant steric hindrance imposed by the dendron units.

Following this, hole-transporting carbazole dendrons were synthesized and incorporated into the MCP. This design uniquely features both a hole-transporting PTAA backbone and carbazole-based side chains, enabling cooperative charge transport from core to periphery. The resulting carbazole dendronized polymers, CzG1-PTAA and CzG2-PTAA, revealed Mw values of 14.5 and 58.8 kDa, respectively (Figure 7a). Furthermore, employing both Fréchet-type benzyl ether and carbazole dendrons enabled the synthesis of a structurally complex and unique doubly dendronized polymer, CzG1-FG2-PTAA, with an Mw of 18.0 kDa and a well-defined alternating dendron sequence. The DP of the dendronized copolymers, calculated by dividing the absolute Mn obtained from MALS by the molecular weight of the corresponding repeat unit, ranged from 6 to 21 depending on the dendron generation and polymer architecture (Figures 7a,b).
UV–vis absorption spectra of benzyl ether dendron-functionalized PTAAs (FG1–FG3 PTAAs) in CHCl₃ showed a consistent λmax at 378 nm, indicating minimal variation across different dendron generations (Figure 8a). In contrast, the doubly dendronized polymers exhibited a red-shifted λmax at 389 nm, suggesting that the spatial proximity of two dendrons has a more significant effect on backbone conformation than dendron size alone (Figure 8b). Furthermore, CzG-PTAAs exhibited enhanced absorption in the 320–350 nm region, which can be attributed to the higher density of carbazole moieties in their side chains (Figure 8b). All dendronized PTAAs exhibited weak blue fluorescence in THF, with photoluminescence quantum yields (ΦF) ranging from 0.15 to 0.22, measured relative to coumarin (ΦF = 1.0) (Table S4).

DSC analysis of dendronized PTAAs revealed a clear trend of decreasing Tg with increasing dendron size (Figure 8c−e).[57, 67-70] From FG1 to FG3, Tg dropped markedly from 181 °C to 75 °C (Figure 8c,e). A similar trend was observed in CzG1-PTAA, CzG2-PTAA, and the doubly dendronized polymers, where Tg decreased from 154 °C to 102 °C as dendron size or density increased (Figure 8d). Moreover, the influence of dendron distribution was further examined by comparing alternating and random FG1- and FG2-PTAAs (Figures 7b and 8c,e). Alternating copolymers exhibited higher Tg values (181 °C for FG1-PTAA, 115 °C for FG2-PTAA) than their random analogues (173 °C and 108 °C, respectively), highlighting the importance of dendron placement in tuning thermal properties.
Conclusion
In summary, the halide-pair-driven MCP strategy was developed as a one-shot modular platform for the library synthesis of sequence-controlled semiconducting PTAAs and their dendronized derivatives. By carefully selecting halide combinations and employing a Pd G4 BrettPhos precatalyst with an additional SPhos ligand, programmed cascade amination was successfully achieved. Mechanistic investigations revealed the presence of a catalyst-transfer process, although its contribution to sequence control was secondary to that of halide pairing. The versatility of the MCP strategy was demonstrated through the diversity-oriented synthesis of semiconducting PTAAs, including dendronized variants. We anticipate that the general principle of the halide-pair-driven MCP strategy can serve as a broadly applicable and accessible platform for synthesizing sequence-controlled semiconducting polymers.
Acknowledgements
The authors appreciate the financial support of the Mid-Career Research Program (No. RS-2025–00517922), Global–Learning & Academic research institution for Master's,·PhD students, and Postdocs (G-LAMP) Program (No. RS-2023–00285390), Carbon to X Project (No. 2020M3H7A1098281), and the Center for Convergence Research of Neurological Disorders (No. 2019R1A5A2026045), through the National Research Foundation of Korea.
Conflict of Interests
The authors declare no conflict of interest.
Open Research
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.

