

The Diels–Alder Reaction as a Mechanistic Probe for Vibrational Strong Coupling
Graphical Abstract
Vibrational Strong Coupling (VSC) was recently shown to modify chemical reactivity, but it is unclear why. We introduce the Diels–Alder (DA) reaction as a mechanistic probe for some of the most popular hypotheses and provide the first experimental investigation of the DA reaction under VSC by measuring the kinetics of six different DA reactions under various coupling conditions. No significant change in kinetics was observed.

Abstract
Vibrational Strong Coupling (VSC) has recently been reported to alter reaction kinetics. Hypotheses on how it does this have been proposed, but open questions remain regarding the importance of the polarity of the reaction mechanism and of intramolecular vibrational redistribution (IVR), among other factors. We propose the Diels–Alder (DA) reaction as a probe to study chemistry under VSC, owing to the high diversity of its reaction partners. Herein, fixed-width cavities and UV–vis spectroscopy were used to determine the rate constants for the reactions of the diene 1,3-diphenylisobenzofuran (DPIBF) with various dienophiles under different coupling conditions. We investigated the effect of coupling six different solvents and of cooperative coupling of the dienophile through the solvent. Secondly, as the DA reaction can be catalyzed by hydrogen bonding, we investigated how the reaction was influenced by coupling alcohol solvents. Finally, we explored the direct coupling of vibrational modes of the dienophiles, including the stretching mode of the reactive C═C bond. In all cases, no substantial changes to the reaction rate constants were observed among the diverse coupling scenarios explored. This work initiates the use of the DA reaction as a mechanistic platform to understand how VSC changes chemistry and invites further experimental and theoretical studies.
Introduction
In recent years, it was found that optical modes may be coupled to vibrational states when injecting molecules within a resonant optical cavity.[1, 2] From this interaction arises the formation of two new light-matter hybridized states called vibropolaritons (VP + and VP−). This phenomenon is referred to as Vibrational Strong Coupling (VSC, Figure 1a), and was found to influence various chemical reactions by changing their selectivity or kinetics.[2-8] Recent reports of reactions that are not altered by VSC[9-13] as well as numerous efforts to model VSC theoretically[14-18] have started bringing insight into the underlying mechanism of VSC-modified chemistry, which nevertheless remains unclear.

One hypothesis lies in the importance of intramolecular vibrational redistribution (IVR).[19-22] When a molecule absorbs an infrared photon, the energy can be redistributed into lower energy vibrational modes before dissipating into the environment. Although reactivity may be altered by such excitation,[23-26] in many cases IVR dissipates on too fast a timescale (typically picoseconds) for this altered redistribution to yield any observable difference in chemical reactivity.[27-30] VSC is believed to alter this redistribution, thereby leading to non-thermal populations and altered reactivity, but there is currently no definitive evidence supporting this hypothesis.[31]
Another point of view is to consider the ensemble of all the reactions that were studied thus far under VSC. Noteworthily, most of the reactions that have been investigated under VSC are highly polar in nature.[2-8, 32] In other words, they involve a nucleophile and an electrophile and charged or highly polar intermediates and transition states, as opposed to non-polar reactions for which the reaction happens in a concerted fashion. Leaning towards non-polar reactions, published VSC studies are scarce, the first example being that of modified rate and selectivity for the ring-opening of a cyclobutene derivative.[33] The other, and to our knowledge, only other notable exception is the study of ultrafast dynamics of the reaction of CN radicals with chloroform, in which no significant effect of VSC could be found.[9] It is unclear whether the scarcity of published data on less polar reactions is the result of VSC not exerting a strong impact on such reactions or of other factors.
The Diels–Alder (DA) reaction can be performed with a myriad of different reaction partners, ranging from ethene to molecules of much higher structural complexity.[34] Owing to this structural diversity, the mechanisms of Diels–Alder reactions are known to vary between concerted to polar (sequential, Figure 1b).[34-36] Similarly, the rate of IVR is found to increase with the density of states of a given molecule, which depends on the molecular structure of the reaction partners (namely their size and functional group diversity among other factors).[37, 38] Varying reaction partners in the DA reaction could therefore significantly alter both the polarity of the mechanism and the rate of IVR, bringing insight into their respective importance in the context of VSC (Figure 1b).
In view of these insights, we undertook the first experimental investigation of Diels–Alder reactivity under VSC (Figure 1c). The reaction rate of 1,3-diphenylisobenzofuran (DPIBF) with different dienophiles was monitored by UV–vis spectroscopy under pseudo-first order conditions. The reaction was performed under VSC of different solvents, including solvents enabling cooperative coupling[4, 8, 32] of the dienophile, where coupling a band of the solvent results in indirect coupling of a degenerate band of the dienophile. Then, alcohol solvents which are known to accelerate Diels–Alder reactivity through hydrogen bonding were also studied. Additionally, we investigated the direct coupling of different vibrational modes of the dienophile, including the reactive C═C stretch. Finally, we propose interesting DA reaction partners for future investigations which should complement our results and thereby help decipher the mechanism of VSC-modified chemistry.
Results and Discussion
The kinetics were measured using a modified version of a previously reported method.[11] Notable differences as well as details of product analyses and reaction partners are given in this subsection.
Fixed-Width Cavities
In this study, we used a new generation of UV–vis transparent fixed-width cavities[39] made by LioniX International, consisting of nine different sizes (C1 to C9, ∼ 6 to 15 µm). The cavities are composed of an outer silica layer (0.5 mm) onto which an adhesive Cr layer (10 nm), a reflective layer of Au (15 nm) and a passivation silica layer (100 nm) were deposited (more details regarding the cavities can be found in the Supporting Information Sections I and III). Also, at our disposal are 12 µm wafers which are devoid of metal layers and that were used for control experiments (later referred to as “cell”). Three different types of measurements were performed in (i) on-resonance cavities where coupling of potentially important vibrations could be achieved, (ii) off-resonance or detuned cavities and (iii) a cell. The fixed-width nature of these cavities minimizes variations in thickness which were recently found to be a potential source of error in studies on VSC-modified chemistry.[40, 41]
Coupling Assessment
We combined experimental dispersion curves with Transfer Matrix Simulations (TMS) to determine which of the nine sizes of cavities at our disposal were of interest by plotting the steady-state electric field distribution within the cavity. These fixed-width cavities have a threshold of 2000 cm−1 in the infrared region, meaning no coupling could be investigated experimentally at lower wavenumbers owing to the transparence limit of its base material (fused silica). Thus, TMS allowed to theoretically investigate coupling below this threshold (all simulations can be found in Supporting Information Section V). For bands above 2000 cm−1, a standard experimental angle dispersion curve was measured, and concentration-dependence studies were performed when necessary. Either way, each band was determined to be either strongly coupled, detuned or decoupled from the cavity. Figure 2 shows the experimental infrared spectrum of the reaction mixture in THF obtained in this work (Figure 2a), while Figure 2b,c show the TMS as obtained for the region from 2000 to 4000 cm−1 in cavities C6 and C8, respectively. Figure 2b illustrates the example of a strongly coupled case (marked ✓ hereafter), where a clear Rabi splitting suggests the presence of polaritons (VP + and VP−) emanating from the interaction between the 11th cavity mode of C6 and the C─H stretching mode of THF. Figure 2c, on the other hand, shows the example of a weak interaction between the 12th cavity mode of C8 (2) and the C─H stretching mode of THF. Indeed, the small spectral feature 1 clearly indicates an interaction with the cavity mode, but the relative intensities of 1 and 2 rather suggest a detuned scenario (marked (✓) hereafter), i.e., the cavity and vibrational modes are not sufficiently degenerate to result in strong coupling. Finally, decoupled scenarios correspond to the absence of any tangible interaction between the cavity and vibrational modes. All details regarding coupling assessment can be found in Section V of the Supporting Information.

Product Analysis
Detailed product analyses were performed for one example of all different dienophiles used by 1H, 13C, and 2D NMR spectroscopy as well as by HRMS. An NMR yield was obtained by integrating the peaks of the products versus the internal standard (dimethyl sulfone). In all cases, the DA adduct was found to be the main product, including when the dienophile was used in “neat” conditions. All details regarding product analysis can be found in the Section IV of the Supporting Information.
Reaction Conditions
All reactions were performed with 1,3-diphenylisobenzofuran (DPIBF, see Table 1) as it strongly absorbs in the visible range (λmax ≈ 410 nm, ε > 2.104 M−1cm−1).[42] The reactions were performed under pseudo-first order conditions with the dienophile in large excess (10 to several thousand equivalents). Considering its limited stability in various solvents, the DPIBF stock solutions used for each set of kinetic experiments were always prepared using tetrahydrofuran (THF), meaning that for all systems, the composition was at least 10% volume in THF. Of note, a small amount of water (1 µL for each 100 µL) was added into all solvents to avoid important differences in water content from one reaction to another. D1 was used in acetonitrile (ACN) and D2 was used in ethyl acetate (EA) and N,N-dimethylformamide (DMF) to potentially allow the cooperative coupling of the dienophile through the C≡N and C═O stretching bands of the solvent, respectively. For direct coupling, only D3 and D4 were investigated as they are liquids at room temperature (and thus are potential solvents). In contrast, both D1 and D2 are solids at room temperature, and the high concentrations required for their coupling produced a reaction that was too fast to measure using the present method. All details on sample preparation can be found in the Section II of the Supporting Information.
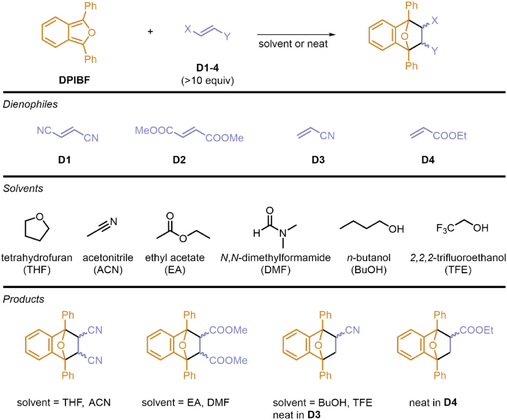 |
Kinetics
For all reactions, a 0.1 M solution of DPIBF (2.7 mg) in THF (100 µL) was prepared. Conversely, a dienophile solution was prepared by mixing the dienophile in the solvent of choice (details on the concentration of the dienophile solutions can be found in Supporting Information Section II). Then, 3 µL of the DPIBF solution were added to 27 µL of the dienophile solution. Quickly after mixing, the reaction mixture was injected into the cavity of choice by means of a micropipette and the cavity was sealed using parafilm. Figure 3 shows the typical UV–vis readout for such a reaction mixture (left, reaction of DPIBF and D1 in THF and cavity C6) as well as the exponential decay of the absorbance of DPIBF which can later be fitted to a first order rate constant (right, more details on obtaining the rate constant are detailed in Section I of the Supporting Information). All kinetic experiments were repeated at least three times, and a standard error was calculated from the different rate constants obtained thereby. Information about the number of replicates in each case as well as the individual rate constants and standard errors calculated therefrom can be found in section VI of the Supporting Information. The molar extinction coefficient of DPIBF was not found to vary significantly between cell and C9 conditions and to agree with values reported in the literature (see Section VII of the Supporting Information),[42] serving as an “in-cavity” calibration of sorts. A conversion of at least 70% was obtained for all kinetic runs, which corresponded to differences in absorbance values of at least ∼0.1 throughout the kinetic run (depending on the reaction speed, and the path length of the cavity). In all cases, including those where the absorbance variation was as low as ∼0.1, we were able to obtain an excellent exponential fit (R2 > 0.999) and the rate constant obtained was found to be independent of the wavelength that was selected (see Section VIII of the Supporting Information).

Effect of Coupling the Solvent on Reaction Kinetics
We started by investigating the effect of coupling different solvents on the reactions. First, the rate constant of the reaction was evaluated in THF with fumaronitrile (D1). Based on our simulations, we identified that the C─O stretching mode of THF (1070 cm−1) was coupled within both cavities C6 and C8. We then checked experimentally whether the other band of interest of THF, the C─H stretching mode (2975 cm−1), could be coupled in either cavity. Doing so, we found C6 to be on-resonance with that vibrational mode. In the case of C8 no polaritonic features were observed, but the simulations evidenced an interaction between the cavity and the vibrational mode, corresponding to a detuned scenario. We thus performed the kinetic measurements within these two cavities and in cell conditions (i.e., a cavity without gold mirrors—and thus no coupling). For all three scenarios, no significant difference in rate constant was observed (Figure 4). For this example and all following examples, all kinetic data can be found in Section VI of the Supporting Information.

We moved on to study solvents for which the direct coupling of a vibrational stretch would result in the cooperative coupling of the dienophile (marked as ✓(c)). For D1, we performed the reaction in ACN for cooperative coupling of the C≡N stretch. Similarly, for dimethylfumarate (D2), we used EA and DMF for cooperative coupling of the C═O stretch. For all three solvents, no substantial difference in rate constant was found, regardless of the cooperative coupling of the C═O and C≡N stretches of the dienophiles (Figure 5). Coupling other bands of the solvents also did not strongly influence the kinetics of the reaction.

Lastly, we turned our attention to alcohols. As previously stated, alcohols are known to catalyze the DA reaction by hydrogen bonding of the dienophile.[43-45] Thus, we considered n-butanol as well as 2,2,2-trifluoroethanol (TFE) as they can both act as a hydrogen bond donor. Starting with n-butanol, we found a small decrease in rate constant (∼10%) for the reaction of D3 and DPIBF when coupling the O─H and C─H stretches (C6), but the same difference was also observed in off-resonance conditions (C1). In the case of TFE, coupling the O─H stretch had no impact on the observed rate constant for the same reaction (Figure 6). The coupling of the OH stretch was determined experimentally by measuring angle dispersion curves as well as by conducting concentration dependence studies. In this specific case, the simulations did not predict the coupling of the OH stretch. This is likely due to lower accuracy in the calculation of the local refractive index, owing to the strong intensity of the OH stretch of both alcohols.

Effect of Coupling the Dienophile on Reaction Kinetics
We were then interested in coupling one of the reaction partners directly. Considering our pseudo-first order conditions, DPIBF was far from being concentrated enough to be coupled. On the other hand, acrylonitrile (D3) and ethyl acrylate (D4) being relatively unreactive, “neat” conditions where the dienophiles were used as the main solvent still yielded reaction times that could be measured conveniently. In these situations, we found that it was possible to couple the C≡N stretch as well as the C─H bending mode of D3, but no substantial change in rate constant was observed compared to the off-resonance cavity and the cell control (Figure 7, left). In both cases, our simulations reported detuned interaction between the cavity and the C─H rock vibration (∼1090 cm−1). Finally, in the case of D4, the C═O and C═C stretches could be coupled, but again no significant difference in observed rate constant was found when comparing the different coupling scenarios (Figure 7, right). Other bands, including the C─H bend, C─H rock, and C─O stretch could be coupled or detuned, but no consistent relationship between their coupling and the rate constant was observed. Of note, the reaction of DPIBF with D3 and D4 in “neat” conditions performed outside the cavity led to the appearance of the exo product as a minor diastereomer, as confirmed by NMR spectroscopy. Unfortunately, inside the cavity, the small quantity of material did not allow for further analysis of the diastereoselectivity, owing to the low concentration (≈ 2.7 mM) of the diene and the small cavity volume (≈ 1 µL).

Discussion
Taken together, these results indicate that there is no substantial VSC-induced modification of the rate constant for the DA reactions tested. The DA reaction is well-known to be highly sensitive to the properties of its environment. For instance, the solvent polarity, which was briefly thought to be modified under VSC,[40, 46] is known to strongly influence the DA reaction.[47, 48] Thus, our results indirectly suggest that there is no significant change in the solvent properties that may affect the DA reaction for all the coupling scenarios we investigated.
As stated in the introduction, reports of reactions for which the mechanism is at least partially concerted (that is, that do not involve a clear nucleophile/electrophile pair) and that are modified by VSC are scarce.[9, 33] Perhaps the closest reaction to have been described is the ring-opening of a cyclobutene derivative, of which the stereoselectivity could be altered by VSC.[33] With these results in mind, it might appear somewhat surprising that the reactions studied herein are not modified by VSC. From an organic chemistry point of view, it should however be noted that the work on the cyclobutene ring-opening reaction significantly differs from this work in that (i) it studies a unimolecular process, (ii) it requires significant heating (90 °C) and (iii) it uses a completely different Fabry–Pérot cavity setup. These key differences coupled to the different phenomenology observed might help to deconvolute the reasons why VSC modifies reactivity.
Recently, pioneering work showed that cooperative strong coupling of the solvent could change the rate of ester cleavage,[32] though it is reportedly difficult to reproduce for reasons that remain unclear.[13] The absence of consequences of cooperative strong coupling for the DA reaction, which features a less polar mechanism than ester cleavage, may contribute to this discussion.
Considering the numerous reports of VSC-modified self-assemblies and reactions that potentially involve hydrogen bonding[3, 5, 49, 50] we investigated the effect of coupling the O─H stretch of alcohol solvents, which are directly involved in these non-covalent interactions. Our results show that such coupling does not have a strong effect on the kinetics of this DA reaction. This observation is in contrast with the recent theoretical reports of altered dynamics of hydrogen bonding under VSC[51, 52] and suggests that these changes do not modify the static properties of hydrogen bonding, and thus, hydrogen bonding catalysis.
Perhaps more surprisingly, we observe that the direct coupling of the dienophile, including the stretching mode of the reactive C═C bond of D4, does not result in any substantial rate change. The direct and selective coupling of the reactive bond is not always achievable, and it is thus interesting to see that, even in this case, the kinetics of the reaction were not impacted.
Besides kinetics under VSC, our work also shows that assessing coupling in VSC studies can be quite challenging. Indeed, in many cases, our simulations evidence that the experimental spectrum that is measured is in fact the complex sum of many non-negligible interactions between the cavity and the reaction mixture (see Section V of the Supporting Information). This once again shows the importance of extensive coupling studies in any VSC study, including through theoretical methods to avoid errors in interpretation. Figure 8 shows the example of the coupling of the reaction mixture of DPIBF and D2 in DMF and cavity C7 between 1000 and 2000 cm−1. Although at first glance, all bands in the simulated spectra might appear strongly coupled, the electric field distribution plot reveals that the spectral features 1 and 2 instead result from a double weak interaction between the vibrational mode and the 6th and 7th cavity modes of C7. It would be challenging to arrive at this conclusion without simulating the field distribution inside the cavity.

As stated in the introduction, the DA reaction can feature structurally diverse dienes and dienophiles. As the kinetics in this work were measured by means of UV–vis spectroscopy, we were limited herein to DPIBF which is highly colored and, concomitantly, to dienophiles which reacted within 5 to 60 min with said diene. The dienophiles used in this work are (i) structurally simple, which would imply that the dissipation of IVR could be slow and thus give time for VSC to operate on the reaction; (ii) electrophilic, which implies that their DA reactivity is at least partially polar (for instance, D3 was found to lie at the non-polar/polar frontier).[36] In both of these aspects there thus remains a significant chemical space to investigate at lower or higher polarity and IVR dissipation rates.
For instance, reactions involving even simpler dienophiles such as ethene would be highly interesting to study, for example through gas-phase VSC.[53] In order to maximize the effect of VSC, slowing down the rate of IVR is not the only option. Studying faster reactions could also bring reaction and IVR rates closer together and increase the probability that the reaction would occur while the populations are altered by VSC. In that context, measuring reliable ultra-fast kinetics could thus be of high interest.[9, 10] Similarly, the recent finding that NMR may be used in the context of VSC[39] could also dramatically increase the scope of dienes and dienophiles that could be studied under VSC, since not all are easily monitored by UV–vis spectroscopy. Finally, owing to the reaction conditions used in this work, VSC of the diene was not possible, thus inviting future studies where the diene can be coupled.
Conclusion
In this work, we took advantage of our recently developed methodology for the measurement of high-quality kinetics under VSC to perform the first-to-date experimental study of the Diels–Alder (DA) reaction under VSC. We selected 1,3-diphenylisobenzofuran (DPIBF) as a diene substrate owing to its strong yellow color and studied its reactivity in eight different sets of conditions. We first investigated the coupling of different solvents, as the DA reaction is well-known to be strongly dependent on solvent properties. Coupling solvents had no substantial impact on the rate of the systems studied, indirectly suggesting that VSC does not modify properties such as solvent polarity or hydrogen bonding ability. Furthermore, cooperative coupling was not found to exert a strong impact on the DA reaction. Finally, we investigated the direct coupling of the dienophile component and found that, even when directly and selectively coupling the stretching mode of the reactive C═C bond, no difference in rate constant was observed. We hope that our results constitute the starting point of a community-wide investigation of the DA reaction as a probe for understanding VSC-modified chemistry.
Supporting Information
The authors have cited additional references within the Supporting Information.[11, 39, 54]
The contents of the Supporting Information are: general information, details on sample preparation and fixed-width cavities, details on product analyses, coupling assessments and finally all kinetic data (spectra, traces, individual rate constants and standard errors).
Acknowledgements
The authors would like to thank Cyril Antheaume (ISIS Strasbourg) for HRMS analyses, as well as Robert J. Mayer (TUM Munich) for fruitful and valuable discussion and Valentin Bauer (ISIS Strasbourg) for the design of the 3D-printed parts of the UV–vis setup. C.M. and M.P. thank the CSC Graduate School, funded by the French National Research Agency (CSC-IGS ANR-17-EURE-0016). The work was also supported by the Interdisciplinary Thematic Institute ITI-CSC via the IdEx Unistra (ANR-10-IDEX-0002) within the program Investissement d'Avenir.
Conflict of Interests
The authors declare no conflict of interest.
Open Research
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.

