

Enantioselective Dearomatization of Pyridinium Salts by Copper-Catalyzed C4-Selective Addition of Silicon Nucleophiles
Graphical Abstract
A C4-selective addition of soft copper-based silicon nucleophiles to pyridinium triflates allows for their enantioselective deraromatization in high yields and with excellent enantioselectivities (see scheme; (R,R)-Ph-BPE=1,2-bis[(2R,5R)-2,5-diphenylphospholan-1-yl]ethane). The thus-formed α-chiral silyl unit can be further used to control the stereoselectivity in subsequent transformations.

Abstract
A copper-catalyzed C4-selective addition of silicon nucleophiles released from an Si−B reagent to prochiral pyridinium triflates is reported. The dearomatization proceeds with excellent enantioselectivity using Cu(CH3CN)4PF6 as the precatalyst and (R,R)-Ph-BPE (1,2-bis[(2R,5R)-2,5-diphenylphospholan-1-yl]ethane) as the chiral ligand. A carbonyl group at C3 is required for this, likely acting a weak donor group to preorganize and direct the nucleophilic attack towards C4. The resulting 4-silylated 1,4-dihydropyridines can be further converted into functionalized piperidine derivatives.
The field of dearomatization reactions has matured tremendously in recent years.1 Two-dimensional molecules can now often be reliably transformed into sp3-rich, three-dimensional scaffolds with high levels of regio- and enantiocontrol.2 An attractive target is the pyridine core as its dearomatization leads to pharmaceutically relevant dihydropyridines3 and, after further functionalization, synthetically valuable piperidine building blocks.4 As an alternative to the at-the-time challenging Birch reduction of pyridines, a reductive 1,4-bissilylation was developed half a century ago.5 It was later realized that the resulting 1,4-bissilylated 1,4-dihydropyridine could be decorated with additional substituents prior to desilylative rearomatization, thereby enabling the regioselective, formal electrophilic substitution of pyridines by temporary dearomatization.6 Given the electron-deficient nature of pyridines and pyridinium ions, the addition of nucleophiles is another possibility to break their aromaticity.7 It was Comins to make use of this strategy,8 and his laboratory also accomplished the C2-selective silylation of acylpyridinium ions bearing a chiral auxiliary at the nitrogen atom (d.r.=98 : 2 using Ph3SiMgX ⋅ 2LiX w/ X=Cl and Br; not shown).9 Diastereoselective additions of copper-based silicon nucleophiles to C4 of acylpyridinium ions with a chiral group at C3 were subsequently achieved by Mangeney10 as well as Comins11 (Scheme 1, top). A catalytic asymmetric version of this transformation is however still missing. Our laboratory has had a long-standing interest in copper-catalyzed silylation reactions employing Si−B reagents as silicon pronucleophiles,12 including their enantioselective 1,2-addition to C=N acceptors.13 A different approach relying on a Pd(0)/Pd(II) catalysis was disclosed by Ohmura and Suginome (Scheme 1, middle).14, 15 Moreover, a few examples of related enantio- and C4-regioselective additions of various carbon nucleophiles were reported recently.16, 17, 18 We therefore decided to investigate the C4-selective dearomatization of prochiral pyridinium ions by copper-catalyzed addition of silicon nucleophiles (Scheme 1, bottom).

Dearomatization of pyridinium ion and pyridine derivatives by silylation. A*=chiral auxiliary, EWG=electron-withdrawing group, X−=counteranion.
We began our study with N-methylpyridinium triflate (1 a) as the model substrate and Suginome's Si−B reagent 2 a (Table 1). A screening of chiral ligands revealed that (R,R)-Ph-BPE (gray box) provides the highest levels of yield and enantioselectivity for the 4-silylated 1,4-dihydropyridine 3 aa (see Table S1 in the Supporting Information). Several copper salts were used to catalyze this reaction in a 4 : 1 1,4-dioxane-H2O solvent mixture (entries 1–4), and Cu(CH3CN)4PF6 emerged as the best (entry 4). We then investigated the influence of the co-solvent and found lower enantioselectivity in THF, Et2O, and toluene (entries 5–7). When replacing NaOtBu by LiOtBu as the base, product 3 aa was obtained in 75 % yield and with good 91 % ee (entry 8). Based on this result, we conducted a series of further optimization reactions using various loadings of Cu(CH3CN)4PF6 and (R,R)-Ph-BPE (see Table S5 in the Supporting Information). From this, the use of 10 mol % of Cu(CH3CN)4PF6 and 20 mol % of (R,R)-Ph-BPE was established as optimal with an enantiomeric excess of 96 % (entry 9). While the 1 : 2 copper-to-ligand ratio secured high enantioinduction, a ratio of 1.0 : 1.2 would still lead to 85 % ee with the yield in the same range (entry 10). We also probed the known counteranion effect, previously described by Karimov and co-workers for a rhodium-catalyzed 1,2-arylation.19 And indeed, OTf− was markedly superior to I−, PF6−, and BF4− in terms of both yield and enantioselectivity (entries 11–13). We find it important to emphasize that all of these reactions were remarkably fast, being completed within a minute or less. The presence of water was critical for this. Repeating the reaction under the optimized procedure in pure 1,4-dioxane had a dramatic effect: The yield was low even after maintaining the reaction overnight, and the ee value was also significantly reduced (entry 14 versus entry 9). We believe that the role of water is two-fold. It solubilizes the pyridinium salt and facilitates the transmetalation step. We also tested zinc- and magnesium-based silicon nucleophiles20 under the non-aqueous conditions but there was no product formation (not shown). The blank control experiments showed that the alkoxide base is essential, even enabling product formation wihout a copper precatalyst15 (entries 15 and 16); the yield was 42 % with no added ligand. Cognate acylpyridinium salts were totally unreactive (not shown).
|
||||||
Entry |
X− |
precatalyst |
base |
co-solvent |
yield [%][b] |
ee [%][c] |
|---|---|---|---|---|---|---|
1 |
OTf |
CuI |
NaOtBu |
1,4-dioxane |
76 |
63 |
2 |
OTf |
CuBr |
NaOtBu |
1,4-dioxane |
52 |
69 |
3 |
OTf |
Cu(acac)2 |
NaOtBu |
1,4-dioxane |
43 |
77 |
4 |
OTf |
Cu(CH3CN)4PF6 |
NaOtBu |
1,4-dioxane |
70 |
85 |
5 |
OTf |
Cu(CH3CN)4PF6 |
NaOtBu |
THF |
21 |
23 |
6 |
OTf |
Cu(CH3CN)4PF6 |
NaOtBu |
Et2O |
20 |
53 |
7 |
OTf |
Cu(CH3CN)4PF6 |
NaOtBu |
toluene |
18 |
0 |
8 |
OTf |
Cu(CH3CN)4PF6 |
LiOtBu |
1,4-dioxane |
75 |
91 |
9[d] |
OTf |
Cu(CH3CN)4PF6 |
LiOtBu |
1,4-dioxane |
75 |
96 |
10[e] |
OTf |
Cu(CH3CN)4PF6 |
LiOtBu |
1,4-dioxane |
76 |
85 |
11 |
I |
Cu(CH3CN)4PF6 |
LiOtBu |
1,4-dioxane |
24 |
15 |
12 |
PF6 |
Cu(CH3CN)4PF6 |
LiOtBu |
1,4-dioxane |
63 |
71 |
13 |
BF4 |
Cu(CH3CN)4PF6 |
LiOtBu |
1,4-dioxane |
37 |
41 |
14[f] |
OTf |
Cu(CH3CN)4PF6 |
LiOtBu |
1,4-dioxane |
40 |
82 |
15 |
OTf |
Cu(CH3CN)4PF6 |
– |
1,4-dioxane |
no reaction |
– |
16 |
OTf |
– |
LiOtBu |
1,4-dioxane |
18 |
0 |
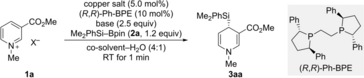
- [a] Unless otherwise noted, all reactions were performed on a 0.10 mmol scale in a 4 : 1 solvent-H2O mixture (1.25 mL) at room temperature under argon atmosphere. [b] Determined after flash chromatography on silica gel. [c] Determined by HPLC analysis on a chiral stationary phase. [d] Cu(CH3CN)4PF6 (10 mol %) and (R,R)-Ph-BPE (20 mol %) used. [e] Cu(CH3CN)4PF6 (10 mol %) and (R,R)-Ph-BPE (12 mol %) used. [f] The reaction was performed in the absence of water and was maintained at room temperature overnight. (R,R)-Ph-BPE=1,2-bis[(2R,5R)-2,5-diphenylphospholan-1-yl]ethane, Tf=trifluoromethanesulfonyl.
With the optimized reaction conditions in hand (entry 9), we tested whether the methodology extends to other 3-substituted pyridinium triflates (Figure 1). The corresponding methyl ketone reacted in lower but still acceptable yield with 90 % ee (4 a→5aa). Conversely, a primary and a tertiary amide as stronger donor groups were not compatible, affording the 1,4-dihydropyridine in poor to moderate yields and with poor ee values (6 a→7aa and 8 a→9aa). No reaction was seen with neither a cyano, a fluoro nor a nitro group (10 a–12 a; gray box). The linear geometry of the nitrile renders a directing effect unlikely, and the pronounced electron-withdrawing nature of these groups could hamper an interaction of the silylcopper(I) nucleophile with the highly electron-deficient pyridinium core in 10 a–12 a.19

Variation of the substituent at C3 of the pyridine core.
We therefore continued with nicotinic acid derivatives for further assessment of the reaction scope (Scheme 2). Systematic variation of R1 in 1 b–h showed that alkyl, benzyl, aryl, and allyl esters are well tolerated (3 ba–ha; Scheme 2A). An additional substituent at C5 as in 1 i and 1 j led to a diminished yield and in one case to lower enantioselectivity (3 ia and 3 ja; Scheme 2B); alkyl groups at C5 thwarted the 1,4-addition with no 1,2-adduct being formed (not shown). In turn, additional substitution at C6 as in 1 k–n was fully compatible with the protocol, affording (hetero)aryl-substituted 3 ka–na in good yields and with consistently high ee values (Scheme 2C). The quinoline skeleton gave the product 3 oa in 85 % yield with 25 % ee. Of note, pyridinium triflates with substitution at C2 (Me, Cl, or OMe) did not react (not shown). A broad range of alkyl and benzyl groups at the nitrogen atom could be introduced (1 p–r→3pa–ra and 1 s–v→3sa–va; Scheme 2D). The functional-group tolerance was generally excellent as further exemplified by 1 w→3wa with a protected indole unit and 1 x→3xa with a C(sp3)−Cl bond. The majority of these 1,4-dihydropyridines is rather sensitive and requires rapid purification. As a result, a few NMR spectra contain minor impurities, e.g. 3 ga, 3 ha, and 3 wa (see the Supporting Information).

Scope of the asymmetric C4-selective silylation of pyridinium triflates derived from nicotinic acid. All reactions were performed with 0.10 mmol of the pyridinium salt 1, 1.2 equiv. of the Si−B reagent 2 a, 10 mol % of Cu(CH3CN)4PF6, 20 mol % of (R,R)-Ph-BPE, and 2.5 equiv. of LiOtBu in 1,4-dioxane (1.0 mL) and H2O (0.25 mL). Yields are isolated after purification by flash chromatography on silica gel. Enantiomeric excesses were determined by HPLC analysis on chiral stationary phases. [a] A 69 % yield with 90 % ee was obtained on a 1.0 mmol scale. Bn=benzyl.
As in several previous copper-catalyzed silylation reactions,12 Me2PhSi−Bpin (2 a) was by far superior to other Si−B reagents (Figure 2). It was only Et3Si−Bpin (2 b) that gave the 1,4-dihydropyridine 3 ab in low yield with a poor ee value. Bulky tBuPh2Si−Bpin (2 c) led to a complex reaction mixture, and Ph3Si−Bpin (2 d) did not react at all.

Variation of the Si−B reagent.
Based on the experimental data about the role of the carbonyl group at C3 (see Figure 1) and key mechanistic insight by Harutyunyan and co-workers,16c, 17e we suggest the plausible catalytic cycle outlined in Scheme 3. The copper(I) precatalyst and bidentate (R,R)-Ph-BPE form the catalytically active silylcopper(I) complex I in the presence of an alkoxide base and water by transmetalation with the Si−B reagent 2. The carbonyl group in 1 not only activates the pyridinium ion but also acts as a directing group to give the weak Lewis pair II. This adduct is likely in equilibrium with another weak adduct, namely III with coordination of the copper(I) center by the pyridine ring between C3 and C4. The (R,R)-Ph-BPE ligand could be monodentate in preorganized III, and the dangling phosphorus donor could then assist the delivery of the soft silicon nucleophile to C4 to eventually yield IV.17e Dissociation of the cationic (R,R)-Ph-BPE-copper(I) fragment in IV releases the dihydropyridine 3 and the copper(I) catalyst. That step involves an anionic oxygen nucleophile in form of either hydroxide (from water), the alkoxide, or triflate and is likely accelerated in the presence of water. Either of these resulting neutral copper(I) complexes engages in another transmetalation with the Si−B reagent 2 to close the catalytic cycle.

Proposed catalytic cycle. Si=triorganosilyl.
To demonstrate the utility of the reaction products, we carried out several transformations of 3 aa (Scheme 4). The electron-rich double bond in 3 aa could be chemoselectively hydrogenated (3 aa→13).21 Further reduction of 13 with NaCNBH3 yielded the piperidine derivative 14 with excellent diastereoselectivity.22 The absolute (and relative) configuration was assigned at this stage. The tertiary amine 14 was converted into the ammonium salt 14 ⋅ HOTf by treatment with MeOTf containing residual water; direct treatment of 14 with HOTf led to decomposition while the corresponding HCl salt did not yield single crystals suitable for X-ray diffraction. The molecular structure of 14 ⋅ HOTf was eventually determined, and the configuration is 3S,4S.23 Dilman's enamine fluorocyanation24 also worked to give the densely functionalized piperidine 15 as a single diastereomer. The enamine in 3 also engaged in a 1,3-dipolar cycloaddition upon reaction with ethyl chlorooximidoacetate (16) to afford the heterocycle 17 in good yield with the shown relative configuration.25 Of note, any attempts to oxidatively degrade the C(sp3)−Si bond in these piperidine derivatives to a secondary alcohol have been fruitless so far.

Derivatization of a dearomatization product. Yields are isolated after purification by flash chromatography on silica gel. Enantiomeric excesses were determined by HPLC analysis on chiral stationary phases. NFSI=N-fluorobenzenesulfonimide.
In summary, we have demonstrated an efficient methodology for the asymmetric synthesis of 4-silylated 1,4-dihydropyridines with excellent enantioselectivities of up to 99 % ee. The approach is based on the Cu−O-mediated transmetalation of Si−B reagents as silicon pronucleophiles.12 The thus-generated copper-based silicon nucleophiles then add to activated pyridinium triflates with superb regioselectivity. The carbonyl group at C3 directs the addition of the silicon nucleophile to C4. The resulting 4-silylated 1,4-dihydropyridines are valuable synthetic intermediates.
Acknowledgments
This work was supported by the China Scholarship Council through predoctoral fellowships to Y.X. (2023–2026) and Z.-Y.Z. (2020–2024). M.O. is indebted to the Einstein Foundation Berlin for an endowed professorship. Open Access funding enabled and organized by Projekt DEAL.
Conflict of Interests
The authors declare no conflict of interest.
Open Research
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.

