

Iron-Catalyzed C−H Alkylation/Ring Opening with Vinylbenzofurans Enabled by Triazoles
Graphical Abstract
An unprecedented C(sp2)−O scission enabled annulations of 2-vinylbenzofurans through a versatile C−H/C−O/N−H functionalization cascade. This reaction was achieved using a peptide-isosteric triazole and an inexpensive, non-toxic iron catalyst.

Abstract
We report an unprecedented iron-catalyzed C−H annulation using readily available 2-vinylbenzofurans as the reaction pattern. The redox-neutral strategy, based on cheap, non-toxic, and earth-abundant iron catalysts, exploits triazole assistance to promote a cascade C−H alkylation, benzofuran ring-opening and insertion into a Fe−N bond, to form highly functionalized isoquinolones. Detailed mechanistic studies supported by DFT calculations fully disclosed the manifold of the iron catalysis.
Transition metal-catalyzed C−H functionalizations nowadays represent an established step- and atom-economical tool in synthetic organic chemistry.1 This approach is thoroughly exploited both in academia and industry,2 for the synthesis of bioactive molecules, applications in material science, optoelectronic and many others.3 Iron, being abundant with lower cost and toxicity, offers the most sustainable applications among other transition metals.4 This pertains to leveraging the desirable iron catalysts for developing novel catalytic transformations, particularly C−H functionalizations.5 Despite the advances in organometallic iron-catalyzed C−H activations, the portfolio of chemical transformations catalyzed by this metal is still quite restricted.6 Thus, the quest for novel reaction patterns is in high demand.
Olefins are abundant feedstock widely employed for transition metals-catalyzed C−H alkylations/annulations.7 These reactions, that constitute one of the most established approaches to forge novel C−C/C−Het bonds, are typically empowered by a migratory insertion of the olefin into a cyclometalated species,8 with the product selectivity being determined by the choice of reaction parameters (Scheme 1, a).

Design of cascade annulations enabled by iron-catalyzed C−H alkylation/ring opening with vinylbenzofurans.
Parallelly, benzofurans are an important class of molecules commonly found in natural products or bioactive compounds.9 The synthetic accessibility of these heterocycles and their targeted functionalizations, contributed to their use as key building blocks in medicinal chemistry and drug discovery.10 Among the synthetic transformations of benzofurans, metal-catalyzed ring-opening reactions are the most versatile as they open access to valuable, highly complex phenol derivatives (Scheme 1, b).11 However, these reactions are challenging, because of the inherent stability of the high-energetic C−O bond12 and often limited in terms of applicability.
Inspired by these concepts, we reasoned to employ 2-vinyl benzofurans as reaction patterns for iron-catalyzed C−H functionalizations. Their unique reactivity, in the presence of iron catalysts, enabled a redox-neutral, cascade sequence for the synthesis of highly functionalized isoquinolones. Notable features of our devised protocol are: i) a C−H alkylation via migratory insertion; ii) a challenging β-O elimination from a C(sp2)−O bond for the ring opening of the benzofuran moiety; iii) a final insertion into the Fe−N bond for the synthesis of the isoquinolone core (Scheme 1, c).
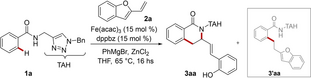
At the outset, inspired by previous findings on triazole-assisted iron-catalyzed C−H alkylations with olefins,13 we probed benzamide 1 a under several reaction conditions in the presence of 2-vinylbenzofuran 2 a. Noteworthily, iron catalysis enabled a cascade reaction leading to the formation of a complex isoquinolone derivative 3 aa (Table 1). The 2-(prop-1-en-1-yl)phenol pendant, formed by the ring-opening of the benzofuran ring, was generated at the C3 position of 3 aa with complete E-selectivity. The optimal reaction conditions devised for this transformation required a bidentate diphosphine ligand with a rigid, aromatic backbone, such as dppbz, PhMgBr (3.0 equiv) and ZnCl2 (2.0 equiv). The use of several iron(II) and iron(III) salts in place of Fe(acac)3 led not only to lower yields but also to the formation of by-product 3’aa (entries 2, 3). In sharp contrast, other transition metals, such as cobalt, nickel or palladium did not provide any transformation under otherwise identical, reaction conditions (entry 4), thus highlighting the unicity of the iron catalytic regime. Among the ligands tested, 2,2’-bipyridine failed to promote any transformation (entry 5). The effect of the backbone of the diphosphine ligands was found to be rather negligible (entries 6, 7). Other organomagnesium reagents, previously found successful in promoting iron-catalyzed C−H activations,14 were not applicable (entries 8, 9). Finally, the role of zinc was investigated. Hence, the presence of zinc is crucial to enable the reactivity of iron catalysts, while its use as a TMEDA adduct was detrimental (entries 10, 11).15
|
||
Entry[a] |
Variation from standard conditions |
3 aa+3’ aa [%][b] |
|---|---|---|
1 |
none |
76 (72)+trace |
2 |
FeCl2 instead of Fe(acac)3 |
30+8 |
3 |
FeCl3 instead of Fe(acac)3 |
37+14 |
4 |
CoCl2 or Ni(acac)2 or Pd(OAc)2 instead of Fe(acac)3 |
– |
5 |
2,2’-bipy instead of dppbz |
– |
6 |
dppen instead of dppbz |
53 (49)+trace |
7 |
dppe instead of dppbz |
50 (45)+trace |
8 |
TMSCH2MgBr instead of PhMgBr |
– |
9 |
i-PrMgBr instead of PhMgBr |
– |
10 |
No ZnCl2 |
– |
11 |
ZnCl2⋅TMEDA instead of ZnCl2 |
– |
- [a] Reaction conditions: 1 a (0.2 mmol), 2 a (0.6 mmol), [Fe] (0.03 mmol), ligand (0.03 mmol), RMgBr (0.6 mmol), ZnX2 (0.4 mmol), THF (0.5 mL). [b] Yields determined using 1,3,5-trimethoxybenzene as the internal standard. In parenthesis, isolated yields. dppe=1,2-bis(diphenylphosphino)ethane; dppbz=1,2-bis(diphenylphosphino)benzene; dppen=cis-1,2-bis(diphenylphosphino)ethene. TMEDA=N,N,N’,N’-tetramethyl-ethylendiamine.
The role of the triazole directing group, for a long time considered only as a surrogate of the most popular 8-aminoquinoline directing group (8-AQ) was subsequently revealed.16 In fact, the reactivity of 2-vinylbenzofuran was evaluated also in the presence of benzamide 4 a (Scheme 2). The only product delivered under iron catalysis was the 1H-inden-3-amine derivative 5 aa. Particularly, only moderate yields could be obtained using the 2,2’-bipy ligand. This finding highlights the distinct chemoselectivity of triazole-assisted transformations. Furthermore, the triazole directing group attributes to the suitable geometry to promote the ring-opening of the benzofuran moiety.17

8-AQ-assisted, iron-catalyzed C−H annulation with 2-vinylbenzofuran.
With the optimized reaction conditions in hands, we evaluated the applicability of the newly devised iron-catalyzed cascade C−H annulation. To this end, several decorated benzamides 1 were tested (Scheme 3).

Substrate scope for benzamides 1.
Differently substituted methylene tethered triazoles 1 b–e were all applicable with high chemoselectivity, delivering the corresponding isoquinolones 3 ba–ea in good yields. The introduction of a new triazole, functionalized with a terminal olefin as for 1 e, opened access to isoquinolone 3 ea, amenable of further transformations by common chemical methods. Electron-rich groups substituted at the para and meta positions of the benzamide ring, performed generally better with respect to the electron-poor analogues. In the first cases, the formation of by-products 3’ was observed only in traces by the 1H NMR analysis of the crude reaction mixtures. This feature was surprisingly observed also for fluoro- and bromo-containing benzamides 1 l–m. An opposite trend was observed for electronically deactivated substrates. In these cases, variable amounts of by-products (3’ na–pa) were formed and could be isolated. Substitution at the ortho-position hampered the cascade sequence and thus C−H alkylated benzamide 3’ ta could only be formed in low-yield.18 A naphthalene ring was also transformed into the corresponding product 3 ua in good yields, while electron-rich heterocycles displayed a varying manifold. Hence, while the use of the indole derivative led to the formation of both compounds 3 va and 3’ va, thiophene 1 w delivered the sole thieno[2,3-c]pyridone 3 wa in average yields.
Subsequently, a set of diversely decorated 2-vinylbenzofuran derivatives was synthesized and utilized as the reaction pattern for the iron-catalyzed cascade C−H annulation (Scheme 4).

Substrate scope for 2-vinylbenzofurans 2.
When employing 5-substitued 2-vinylbenzofurans 2 b–h, we observed an opposite trend with respect to the one disclosed for benzamide derivatives. Particularly, the catalytic reaction in the presence of 2-vinylbenzofurans decorated with electron-donating groups 2 b–d, led to the formation of both isoquinolones 3 ab–ad and alkylated by-products 3’ab–ad in variable yields. In contrast, when catalytic reactions were performed using 2-vinylbenzofurans functionalized with electron-withdrawing groups 2 f–h, isoquinolones 3 af–ah were the only products that could be isolated from the reaction mixture. Again, the use of fluoro-containing vinylbenzofuran 2 e represented a deviation from this trend since both compounds 3 ae and 3’ae could be forged in the catalytic reaction in overall moderate yields. Substrates with functional groups at other positions of the benzofuran were also synthesized and tested under otherwise identical reaction conditions (see Supporting Information for limitations of the method and mass-balance analysis). A 6-methoxy-2-vinylbenzofuran 2 i reacted smoothly leading to the formation of 3 ai in good yields. Contrarily, a 7-methoxy-2-vinylbenzofuran analogue 2 j turned out to be more challenging. A similar outcome was observed when 5,7-dichloro-2-vinylbenzofuran 2 k was used as the reaction pattern. In all these cases, no formation of by-products 3’ was detected in the reaction mixtures. 2-Vinylnaphtho[2,1-b]furan 2 l turned out to be reactive under the iron catalytic regime. However, 3’ al was the only product delivered, although in fair yield. Finally, biomass-derived 2-vinylfurans 6 were tested under otherwise identical reaction conditions. Hence, the presence of the methyl group was mandatory to promote a challenging cascade C−H annulation leading to compound 7 ab, functionalized with a β,γ-unsaturated ketone pendant. Overall, the developed method displayed a broad and useful substrate scope, thus providing a new approach for non-trivial functionalizations of bioisosteric and pharmacologically-relevant, triazole compounds.19
To gain insights into the working mode of the iron-catalyzed cascade C−H annulation, mechanistic studies were performed next.
Inter- and intramolecular competition experiments using deuterium-labelled compound [D]5–1 b revealed a lack of primary kinetic isotope effect. This outcome suggested that the C−H metalation step is facile (Scheme 5, a and b). Subsequently, compound [D]5–1 a was submitted to the optimal catalytic conditions, leading to the formation of the corresponding product without significant deuterium scrambling at the ortho-position (Scheme 5, c). This finding is supportive of the C−H activation to be irreversible.

Competition experiments and deuterium labelling.
A deuterated 2-vinylbenzofuran [D]n–2 a was synthesized and reacted with benzamide 1 a. Also in this case, isoquinolone [D]n–3 aa was formed in good yields with retention of the deuterium content (Scheme 5, d). Finally, a model reaction was stopped by the addition of DCl, leading to the selective labelling of only one vinylic position (Scheme 5, e). These results are not in agreement with a manifold initiated by oxidative addition of the metal and subsequent olefin hydrometallation.
Intrigued by the unique features of the iron-catalyzed transformation, promoted by the triazole directing group, DFT calculations were conducted at the PW6B95-D4/def2-TZVPP+SMD(THF)//TPSS-D3(BJ)/def2-SVP level of theory (Figure 1). Substrate 1 a and 2-vinylbenzofuran 2 a were chosen to model the iron-catalyzed C−H cascade annulation. Considering the nature of the iron(II) complexes, the reaction pathway was calculated for all possible three spin states: singlet (low-spin), triplet (intermediate-spin), and quintet (high-spin) (Figure 1).20

Computed Gibbs free energy profile (ΔG338.15) in kcal mol−1 for the iron-catalyzed cascade C−H annulation with 2-vinylbenzofuran at the PW6B95-D4/def2-TZVPP+SMD(THF)//TPSS-D3(BJ)/def2-SVP level of theory. Non-relevant hydrogens in the transition state structures were omitted for clarity. Superscripted 1, 3, and 5 represents singlet, triplet and quintet spin states, respectively.
Four elementary steps involved in the cascade C−H annulation were investigated, namely a) C−H activation, b) alkene migratory insertion, c) β-O-elimination, and d) allene migratory insertion. The C−H activation proceeds through a ligand-to-ligand hydrogen transfer mechanism (LLHT)21 with an energy barrier of 18.3 kcal mol−1 (TS(1–2)) to form cyclometalated species Int2.22 The low energy barrier results from an into-play spin-crossover that results in a drastic reduction of the energy barrier when compared to the quintet TS(1–2) surface. Upon olefin coordination, Int3 undergoes migratory insertion through TS(3–4) with an energy barrier of 24.0 kcal mol−1. Here as well, spin crossover was observed from a quintet Int3 to a singlet TS(3–4) surface, giving rise to Int4 in a quintet state.
After the olefin rotation, the Int4’ undergoes β-O-elimination TS(4’–5),23 which revealed to be endergonic with an energy barrier of 28.1 kcal mol−1, generating the η2-allene coordinated Int5. Thereafter Int6 is formed via intramolecular migratory insertion TS(5–6), with an energy barrier of 38.3 kcal mol−1. This suggests that the intramolecular migratory insertion into the N−Fe bond is the rate determining step.
Additionally, Hammett analysis was performed computationally (see Supporting Information) at the same level of theory. The β-O-elimination step was considered for this analysis, identifying it as the point of deviation from the investigated pathway which leads to linear by-products 3’. The careful analysis of the Hammett plots indicated that the more electron-withdrawing meta-and para-substituents at the benzamide substrate may facilitate the formation of linear by-products 3’, lowering the formation of isoquinolones 3. The low chemoselectivity observed for these electron-demanding, more acidic amides could be further attributed to the competitive proto-demetalation24 of Int4 that releases by-product 3’ thus preventing the β-O-elimination step. In contrast, electron-rich substituents were shown to largely favor the formation of isoquinolones 3.
Based on our mechanistic findings we propose a catalytic cycle initiated by a facile C−H activation via LLHT (Scheme 6). Subsequently, coordination of vinylbenzofuran 2 a and a fast migratory insertion, provide the key iron species Int4. This intermediate undergoes a rotation of the side-chain arm that enables an endergonic cis-β-O elimination leading to the Int5. Thereafter, a rate-determining intramolecular insertion into the N−Fe bond forms Int6, that finally release the product 3 aa via proto-demetalation, regenerating the active iron species.

Proposed catalytic cycle.
In conclusion, we disclose the assembly of highly functionalized isoquinolones based on a cascade C−H annulation with 2-vinylbenzofurans enabled by earth-abundant and non-toxic iron catalysts. The versatility of the iron catalysis was demonstrated by several late-stage diversifications of the phenol and triazole motifs. (see Scheme 7 and SI).

Reaction conditions: a) 3 aa (0.2 mmol), pyridine (3.0 equiv), trifluoromethanesulfonic anhydride (1.5 equiv), CH2Cl2 (1 mL), 0 °C, 1.5 hs. b) 8 aa (0.1 mmol), Pd(OAc)2 (5 mol %), PCy3 (6 mol %), phenylboronic acid (1.5 equiv), KF (3.3 equiv), THF (0.3 mL), 50 °C, 5 hs. c) 8 aa (0.1 mmol), Pd(dppf)Cl2 (5 mol %), HCOOH (2.0 equiv), Et3N (5.0 equiv), DMF, 80 °C, 16 hs. d) 8 aa (0.1 mmol), Pd(PPh3)4 (10 mol %), CuI (10 mol %), Et3N (3.0 equiv), Trimethylsilylacetylene (3.0 equiv), CH3CN (0.5 mL), 110 °C, 16 hs. e) 8 aa (0.1 mmol) Pd(OAc)2 (10 mol %), rac-BINAP (15 mol %), Cs2CO3 (2.8 equiv), benzylamine (2.0 equiv), toluene (0.2 mL), 80 °C, 24 hs. f) 3 aa (0.1 mmol), LiAlH4 (4.0 equiv), THF (1 mL), 60 °C, 16 hs. g) 3 ea (0.1 mmol), Grubbs catalyst 2nd generation (10 mol %), methylacrylate (4.0 equiv), CH2Cl2 (1.0 mL), 40 °C, 16 hs. Isolated yields given. Tf=trifluoromethanesulfonyl. TMS=trimethylsilyl
Importantly, the versatile triazole directing group was key to success for a new mode of action for iron catalysts, based on an unprecedented β-O elimination that drives the C(sp2)−O activation of benzofuran heterocyclic moieties. Computational and experimental studies revealed a spin-crossover phenomena and fast C−H activation as key elements of efficient iron catalysis.
Supporting Information
The authors have cited additional references within the Supporting Information.25-35, 36, 37-39
Acknowledgments
The authors thank Centro Interdipartimentale di Misure of the University of Parma for the NMR measurement. This research was supported by National Programs (PON “Ricerca e Innovazione” 2014–2020 Azione IV.4 and Azione IV.5), by the Italian MIUR (PRIN 20227Z3BL8), and by the University of Parma (FIL–Bando di Ateneo per la Ricerca 2022, Azione B). Generous support from the DZHK, the ERC Advanced Grant no.101021358, the DFG Gottfried-Wilhelm-Leibniz-Preis, and SPP 2363 LA are gratefully acknowledged. Open Access publishing facilitated by Università degli Studi di Parma, as part of the Wiley - CRUI-CARE agreement.
Conflict of Interests
The authors declare no conflict of interest.
Open Research
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.

