

Photocatalytic Decarboxylative Functionalization of Cyclopropenes via Cyclopropenium Cation Intermediates
Graphical Abstract
We report a photocatalytic decarboxylative functionalization of cyclopropenes. Starting from cyclopropenyl redox-active esters, easily accessible through the rhodium- or silver-catalyzed cyclopropenation of alkynes, we could access a wide range of diverse cyclopropenes under mild photoredox conditions. The transformation displayed a broad scope and a remarkable functional group tolerance. Mechanistic studies supported aromatic cyclopropenium cations as reaction intermediates.

Abstract
A photocatalytic decarboxylative functionalization of cyclopropenes is reported. Starting from a broad range of redox-active ester-substituted cyclopropenes, cyclopropenylphthalimides can be synthesized in the absence of a nucleophile. Alternatively, different carbon and heteroatom nucleophiles can be introduced. The transformation proceeds most probably through the formation of an aromatic cyclopropenium cation, followed by trapping with the nucleophiles.
Cyclopropenes are the smallest cyclic alkenes. Due to their high ring strain, cyclopropenes possess a unique reactivity compared with other cyclic alkenes, leading to a wide variety of synthetic applications.1 In particular, cyclopropenes represent a versatile platform for the stereoselective synthesis of polysubstituted cyclopropanes through double-bond functionalization.2 The resulting cyclopropanes are conformationally rigid, three-dimensional compounds with well-defined exit vector relationships for the substituents, which makes them highly useful for medicinal chemistry, as exemplified with already 79 approved drugs (Scheme 1A).3 Nevertheless, only a limited amount of substitution patterns on cyclopropenes are currently easily accessible due to the narrow scope of the two main methods used for their synthesis (Scheme 1B). The rhodium and silver-catalyzed cyclopropenation of alkynes with diazo compounds requires in most cases at least one electron-withdrawing group to stabilize the metal carbene intermediate (Scheme 1B, Eq. 1).4 Cyclopropenes without electron-withdrawing groups are usually obtained via multistep sequences, based on elimination reactions (Scheme 1B, Eq. 2). The scope of this approach is limited to 3,3-disubstituted products with few functional groups tolerated due to the harsh basic conditions used.5 Therefore we become interested in developing new synthetic routes towards cyclopropenes.

A: Stereoselective functionalization of cyclopropenes B: Synthetic strategies towards cyclopropenes C: Catalytic methods for CPC generation D: Photocatalytic decarboxylative CPC generation.
Our attention was attracted by the chemistry of cyclopropenium cations (CPCs)—stable aromatic carbonium ions derived from cyclopropenes. Since the first isolation of a CPC in 1957 by Breslow, these compounds were shown to react with nucleophiles resulting in the corresponding cyclopropene products.6 Despite their potential synthetic utility, CPCs were mostly neglected by the mainstream synthetic community due to the inefficient procedures available for their preparation. In 2022, the group of Suero developed a new synthesis of CPCs using diazo-substituted hypervalent iodine reagents (Scheme 1C, Eq. 1).7 A broad scope of nucleophiles could be used, giving access to structurally diverse cyclopropenes. However, only ester-substituted CPCs can be synthesized, as the electron-withdrawing group is required for stabilizing the used hypervalent iodine reagents.
In 2023, Wilkerson-Hill reported another catalytic method for the generation of CPC intermediates by sequential fluoride abstraction from difluorocyclopropenes, resulting in the double addition of nucleophiles (Scheme 1C, Eq. 2).8 In this methodology, the nucleophiles are mostly limited to tetrasubstituted ketene silyl acetals, and double addition is unavoidable. We envisaged that starting from redox-active ester substituted cyclopropenes under photoredox conditions we would be able to generate cyclopropenium cations via oxidation of the intermediate radicals.9 The cations can be then trapped in situ by nucleophiles present in the reaction mixture to give cyclopropenes without an ester substituent. Interestingly, in a seminal communication from Joglar and co-workers, one single example of conversion of a phthalimideoxyester cyclopropene into the corresponding N-(2-cyclopropenyl)phthalimide under sensitized UV irradiation was reported.10 A radical recombination mechanism without the formation of a CPC was proposed, strongly limiting the synthetic applicability of the method. Indeed, only one example of C−H bond formation was demonstrated by hydrogen atom transfer to the intermediate radical. We therefore hypothesized that our mechanistically distinct approach proceeding via a CPC would overcome these strong limitations. In addition, rather than the multi-step procedure applied by Joglar and co-workers, we envisage to synthesize the required starting materials using the rhodium or silver-catalyzed cyclopropenation of alkynes with redox-active ester substituted diazo compounds—reagents introduced by Mendoza and co-workers.11 Herein, we report the successful implementation of this strategy, resulting in a new general synthesis of cyclopropenes via the attack of nucleophiles onto cyclopropenium intermediates (Scheme 1D).
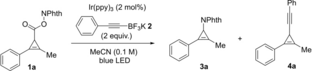
To test our hypothesis we prepared cyclopropene 1 a in 61 % yield by the rhodium-catalyzed cyclopropenation of the corresponding commercially available alkyne (see optimization in the Supporting Information). We then performed a test reaction using alkynyltrifluoroborate 2, an established nucleophile for radical-polar crossover reactions under photoredox conditions (Table 1, Entry 1).12 Instead of the desired product we observed decarboxylative amination product 3 a. Generally, 3-aminocyclopropene derivatives can be generated by addition of nitrogen nucleophiles to cyclopropenium cations,13 by Curtius rearrangement,14 and by elimination from haloaminocyclopropanes.15 However, all these methods require tedious multistep synthetic sequences. A high regioselectivity for nucleophile addition to the less substituted position of the cyclopropenium cation was observed, in good accordance with the literature.16 An experiment without trifluoroborate nucleophile resulted in the recovery of the starting material (Entry 2). We hypothesized, that the addition of a weak Brønsted acid would result in the protonation of the phthalimide anion (pKa=8.3), preventing it from attacking the cation. Indeed, addition of collidinium tetrafluoroborate (pKa=7.43) resulted in the formation of alkynylated product 4 a in good yield (Entry 3). The addition of LiBF4 also led to the formation of 4 a in 74 % yield (Entry 4). Optimization of the photocatalyst showed that using 5 mol % of 1,3-dicyano-2,4,5,6-tetrakis(diphenylamino)-benzene (4DPAIPN) gave the best yield (Entry 5) (see Supporting Information for optimization details). An examination of other acidic additives revealed that acetic acid was also promising (Entry 6). Increasing its amount to 4 equivalents resulted in 4 a in 90 % yield (Entry 7). The addition of a basic additive, in contrast, resulted in the formation of amination product 3 a in 89 % yield (Entry 8) (see Supporting Information for full optimization data).
|
|||
Entry |
Deviation from above conditions |
Yield 3 a[b] |
Yield 4 a[b] |
|---|---|---|---|
1 |
none |
75 % |
– |
2 |
w/o trifluoroborate |
– |
– |
3 |
+CollidiniumBF4 (2 equiv.) |
– |
64 % |
4 |
+LiBF4 (0.5 equiv.) |
– |
74 % |
5 |
4DPAIPN (5 mol %)+LiBF4 (0.5 equiv.) |
– |
84 % |
6 |
4DPAIPN (5 mol %)+AcOH (2 equiv.) |
6 % |
75 % |
7 |
4DPAIPN (5 mol %)+AcOH (4 equiv.) |
– |
90 % |
8 |
4DPAIPN (5 mol %)+Na2CO3 (1.75 equiv.), w/o trifluoroborate |
89 % |
– |
- [a] The reactions were performed on 0.1 mmol scale (see Supporting Information for the detailed reaction procedure). [b] NMR yield, measured using CH2Br2 as an internal standard.
With optimized conditions in hand, we explored first the scope of cyclopropenes (Scheme 2A). From 1 a, amination product 3 a was isolated in 90 % yield. Different substituents in the para position of the aromatic ring were tolerated, including an electron-donating methoxy substituent (3 b), electron-withdrawing substituents (3 c, 3 d), and halogens (3 e–f). The reaction worked well for meta- (3 g, 3 h) and ortho-substituted arenes (3 i). Propyl-substituted product 3 j was obtained in 79 % yield. A protected allylic alcohol was also tolerated (3 k, 47 % yield). A cyclopropyl substituent remained untouched to give 3 l. Benzyl-substituted cyclopropene 3 m was obtained in 43 % yield. Heteroaryl-substituted cyclopropene 3 n was isolated in 74 % yield. Remarkably, cyclopropenes bearing only one alkyl substituent can also undergo decarboxylative functionalization efficiently. To the best of our knowledge, monosubstituted cyclopropenium salts have never been isolated, probably due to their lower stability. These elusive species can apparently still be generated under our conditions and trapped by the nucleophile in situ. Propyl (3 o) and phenethyl- (3 p) substituted aminocyclopropenes were obtained in 81 and 80 % yield. The transformation also tolerated an alkyl chloride (3 q), a protected amine (3 r), a protected alcohol (3 s), a nitrile (3 t), and an ester (3 u) functionality. Cyclopropenes bearing aliphatic cyclic (3 v) and heterocyclic (3 w) substituents also worked well. Tertiary alkyl substituted cyclopropene 3 x can be isolated in 87 % yield. Symmetrical (3 y) and unsymmetrical (3 z) dialkylcyclopropenes were also formed. Aminocyclopropene 3 aa was obtained from a α-D-allofuranose-derived starting material (1 aa). For 21 out of 33 examples, contamination with an inseparable minor regioisomer, resulting from the attack of the nucleophile on the alkyl-substituted carbon of the cyclopropenium ion was below 5 % and therefore could not be quantified by NMR. For 11 substrates, the amount of impurity was between 5 and 10 % and could be quantified.

Scope of the decarboxylative amination of cyclopropenes. A: From 3-monosubstituted cyclopropenes. B: From 3-aryl cyclopropenes. [a] The reactions were performed on a 0.3 mmol scale (see Supporting Information for the detailed reaction procedure). The isolated yield (for the mixture of regioisomers, if applicable) is reported. [b] regioisomer ratio (rr) is determined by analysis of the 1H NMR spectrum of the isolated product; rr is >19 : 1 if not indicated.
Some cyclopropenes could not be obtained through the Rh-catalyzed cyclopropanation: pyridine-substituted cyclopropene 1 ab and cyclopropenes 1 ac and 1 ad bearing a phenyl group and a bulky alkyl substituent. A few substrates were unsuccessful in the photoredox-mediated amination: Cyclopropene 1 ae, containing an allyl chloride functionality, displayed low conversion with no product formation. With cyclopropene 1 af bearing a phthalic anhydride functionality a complex mixture was observed.
As a more substituted substrate class, we then examined the reaction of 3-aryl substituted cyclopropenes 1 ag–1 al (see Supporting Information for cyclopropene synthesis) (Scheme 2B). Starting from cyclopropene 1 ag, product 3 j was obtained in excellent yield. This result allowed us to address some of the limitations observed with the previously investigated class of cyclopropenes. Indeed, sterically hindered cyclopropene 3 ah could be now obtained in 91 % yield from cyclopropene 1 ah. Silyl-substituted aminocyclopropene 3 ai was also isolated in 73 % yield. When considering the reported protodesilylation of silylcyclopropenes,5 1 ai can be considered as the synthetic equivalent of a monoarylcyclopropenium cation. Diarylcyclopropene 3 aj was obtained in 42 % yield. The lower yield in this case can be explained by the low solubility of the product in MeCN, which led to poor light penetration in the reaction mixture. Moreover, some degradation of the product may occur through sensitization of the diarylalkene moiety of 3 aj. The addition of nucleophiles to unsymmetrical trisubstituted cyclopropenium cations usually results in a mixture of regioisomeric products.6a Interestingly, starting from cyclopropene 1 ak we could obtain 3 ak with remarkable regioselectivity. Starting from cyclopropene 1 al a mixture of regioisomers 3 al was formed, with the predominance of the product resulting from the attack of the nucleophile on the alkyl-substituted carbon.17 To demonstrate the scalability of our method, the synthesis of 3 a was performed on a 1 g scale, resulting in the formation of 3 a in 86 % yield.
We then moved to the exploration of the nucleophile scope (Scheme 3). Starting from model substrate 1 a we performed the alkynylation using acetic acid as an additive to give 4 a in 86 % yield.18 Other organotrifluoroborates were then examined. Alkyl-substituted alkynes 4 b–d were obtained in good yields. Alkenylation product 4 e was obtained upon reaction with E-styryl trifluoroborate with a slight erosion of the double bond geometry. Cyclohexenyl-substituted cyclopropene 4 f was also obtained in good yield. Electron-rich (hetero)aryl trifluoroborates could also be used as nucleophiles, giving products 4 g and 4 h. Neutral aryltrifluoroborates appeared to be not nucleophilic enough to undergo the transformation (for unsuccessful nucleophiles see Supporting Information). Allylation of the CPC resulted in the formation of 4 i. Other types of nucleophiles were then examined. The reaction with the TBS enol ether of acetophenone resulted in the formation of 4 j in 78 % yield. The reaction with NaCN afforded cyclopropenyl nitrile 4 k. Aminocyclopropene 4 l was obtained using tert-butyl carbamate as a nucleophile. When P(OEt)3 was used as a nucleophile, phosphonate product 4 m was formed. The lithium salt of dimethylmalonate, which was not compatible with the AcOH additive, could be efficiently introduced using LiBF4 as an additive, giving product 4 n. Unfortunately, different O-nucleophiles (cyclohexanol, phenol, benzoate) were not successful, presumably due to the instability of the products under the reaction conditions (see Supporting Information for a list of unsuccessful nucleophiles).

Nucleophile scope. [a] The reactions were performed on a 0.3 mmol scale (see Supporting Information for the detailed reaction procedure). Isolated yields are reported. [b] The reaction was performed using LiBF4 (0.5 equiv.) instead of AcOH.
We believe that our methodology significantly expands the chemical space of easily accessible cyclopropenes. A systematic study of the reactivity of these new types of cyclopropenes lies beyond the scope of this communication. Nevertheless, we still performed selected synthetically useful transformations on product 3 a (Scheme 4). Reduction of the double bond, followed by removal of the phthalimide protecting group gave aminocyclopropane 6 (Scheme 4a).19 Such compounds are difficult to access by other means due to the cis-stereochemistry of the three substituents on the cyclopropane. Bicyclic compound 8 can be obtained through Pauson–Khand reaction of 3 a with cobalt complex 7 (Scheme 4b).20 Finally, a copper-catalyzed hydroboration gave tertiary cyclopropylboronate 9 as a single diastereoisomer (Scheme 4c).21

Synthetic transformations of 3 a. [a] See Supporting Information for the detailed reaction procedures.
To better understand the mechanism of the transformation we performed several experiments. In the absence of photocatalyst, light or additive the reaction did not take place (Scheme 5a). A Stern–Volmer experiment showed quenching of the photocatalyst fluorescence by 1 a (Ksv=106.9 M−1) (Scheme 5b). When the reaction was performed in the presence of TEMPO, no product was formed (Scheme 5c). A low amount of TEMPO adduct 10 was detected by HRMS and NMR spectroscopy of the crude mixture. When the reaction of cyclopropene 1 o was performed with AcOH as an additive, but without an external nucleophile, cyclopropenylacetate 11 was observed by NMR as a mixture with 3 o. All attempts to isolate 11 were unsuccessful. When alkynyltrifluoroborate 2 was added after completion of the reaction and the mixture was stirred for 3 more hours in the dark, the alkyne 12 was formed in 71 % yield (Scheme 5d). We also confirmed that amination product 3 a (which should be more reactive then 3 o, as the corresponding CPC is more stable) did not react with an external alkyne nucleophile under the reaction conditions (Scheme 5e). Finally, when the decarboxylative amination reaction of 1 a was performed in the presence of methyl-substituted phthalimide 14, a mixture of both possible products (3 a and 15) was formed in almost equal amounts (Scheme 5f).

Mechanistic experiments.
Based on these experiments the following mechanism can be proposed (Scheme 6). Starting material 1 would first undergo a single-electron reduction by the excited-state photocatalyst, resulting in radical-anion I. Then, fragmentation would occur, leading to cyclopropenyl radical II. We believe that Na2CO3 plays a crucial role in this process, allowing an ion exchange between sodium and the 4DPAIPN radical cation, therefore suppressing the back electron transfer to radical anion I.22 Alternatively, the acetic acid additive would protonate the radical anion intermediate, also neutralizing the ion pair. The protonated phthalimide formed after fragmentation is much less nucleophilic, and the formation of the amination product is therefore suppressed. Cyclopropenyl radical II would then be oxidized by the radical-cation of 4DPAIPN, forming CPC III and regenerating the ground-state photocatalyst. When AcOH is used as an additive, this cation can reversibly react with acetate, present in the mixture to form cyclopropenyl acetate IV, which is acting as a cation reservoir. We believe that the formation of acetate IV may lower the concentration of reactive CPC III, thereby preventing unproductive side reactions and increasing the yield of the desired product. Finally, CPC III would react with a nucleophile present in the reaction mixture, resulting in the desired products 3 or 4.

Proposed speculative mechanism.
In conclusion, we have reported a photocatalytic decarboxylative functionalization of cyclopropenes proceeding via the intermediacy of a putative cyclopropenium cation. Cyclopropenylphthalimides could be synthesized in high yield with a broad scope in the absence of external nucleophiles. The use of acetic acid or lithium tetrafluoroborate as additive allowed the addition of a wide range of nucleophiles, resulting in the synthesis of uncommon cyclopropene products, difficult to access by other synthetic methods. A speculative radical-polar crossover catalytic cycle was proposed based on the available literature and the mechanistic studies performed. The investigation of other transformations based on CPC reactivity is currently ongoing in our laboratory.
Supporting Information
The authors have cited additional references within the Supporting Information.23 Raw data for NMR, IR and MS are freely available on the platform zenodo: https://doi.org/10.5281/zenodo.11263698
Acknowledgments
We thank Dr. Xiangdong Li and Julien Borrel for donating some of the compounds, used in this study. We thank the Swiss National Science Foundation (Grant No. 200020_212129) for financial support. Open Access funding provided by École Polytechnique Fédérale de Lausanne.
Conflict of interests
The authors declare no conflict of interest.
Open Research
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.

