

Photochemical Desaturation and Epoxidation with Oxygen by Sequential Flavin Catalysis
Graphical Abstract
In nature, flavoenzymes mediate mechanistically unrelated transformations such as desaturations and epoxidations with oxygen from air. We disclose the combination of both activities in a sequential fashion using molecular flavin catalysts in the organic laboratory. Our protocol consists of initial photochemical desaturation of silyl enol ethers and subsequent α,β-epoxyketone formation with molecular oxygen.

Abstract
Catalytic desaturations are important strategies for the functionalization of organic molecules. In nature, flavoenzymes mediate the formation of α,β-unsaturated carbonyl compounds by concomitant cofactor reduction. Contrary to many laboratory methods for these reactions, such as the Saegusa-Ito oxidation, no transition metal reagents or catalysts are required. However, a molecular flavin-mediated variant has not been reported so far. We disclose a photochemical approach for silyl enol ether oxidation, which leads to α,β-unsaturated ketones (13 examples) in very good yields. The flavin catalysts are stable throughout the desaturation reaction, and we successfully applied them in a subsequent aerobic epoxidation by simply changing the reaction conditions. This protocol allowed us to directly convert silyl enol ethers into α,β-epoxyketones in a one-pot fashion (12 examples). Sequential flavin catalysis is not limited to one specific reactivity combination and can, inter alia, couple the photochemical oxidation with radical additions. We anticipate that flavin-catalyzed desaturation will be applicable to other substrate classes and that its sequential catalytic activity will enable rapid substrate diversification.
Flavoenzymes mediate a fascinating plethora of transformations.1 The unique chemical environment around the flavin cofactor controls its oxidation state and substrate accessibility, allowing it to choreograph many different types of reactions.2 In contrast to the rich enzymatic repertoire, the use of molecular flavin catalysts in organic synthesis is limited.3 In the present study, we were particularly interested in the ability of molecular flavin catalysts to couple two chemically distinct steps in an iterative one-pot procedure, enabling rapid substrate diversification.4 Catalytic desaturation5 and aerobic epoxidation were chosen based on their synthetic utility and reported flavoenzyme activity.
In flavin-dependent desaturase enzymes (Figure 1A), flavin adenine dinucleotide (FAD) accepts a hydride to release the oxidized product concomitant to being reduced to the hydroquinoid state (FADH2). Thiazoline (1, X=S) and oxazoline (1, X=O) desaturation proceed via this mechanism,6 which is also operative in dihydroorotate dehydrogenase. The latter forms orotidine precursor 2 for uridine biosynthesis.7 In the organic laboratory, desaturation adjacent to carbonyl positions is typically achieved by silyl enol ether formation and subsequent Saegusa-Ito reaction (Figure 1B), which requires stoichiometric amounts of a palladium oxidant.8 The reaction is very valuable for natural product synthesis and the preparation of active drug molecules.9 Significant improvements on the original conditions have been reported, including catalytic strategies with a sacrificial oxidant,10, 11 hypervalent iodine reagents,12 photochemical generation of singlet oxygen,13 and electrochemical oxidation.14 However, with the exception of enzyme-mediated examples,15 flavin-catalyzed desaturations are still lacking as a synthetic method.16

Desaturation and epoxidation catalysis. A) Flavoenzyme desaturation. B) Saegusa-Ito reactions in the organic laboratory. C) Flavoenzyme activity in epoxidation with O2. D) Our strategy for combining both activities in sequential catalysis.
In an orthogonal reactivity, flavoenzymes activate molecular O2 from air for oxygenation reactions (Figure 1C).17 These reactions require the reduced cofactor (FADH2) and result inter alia in the formation of oxiranes.18 In the organic laboratory, such reactivity typically requires oxidants such as mCPBA.19 It was the aim of this study to enable molecular flavin-catalyzed desaturation and to achieve sequential epoxidation in a one-pot reaction with the same flavin catalyst. This strategy would enable the direct conversion of silyl enol ethers into α,β-epoxyketones.
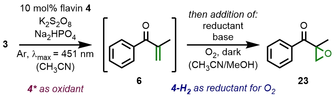
We initiated our studies with silyl enol ether 3 and probed whether molecular flavins serve as oxidants for desaturation in analogy to the enzymatic transformations. However, no thermal reactivity was observed even under elevated temperatures. This shifted our interest to photochemical conditions,20 since excited quinoid flavins are strong oxidants with  (34*/4⋅−)=+1.35 V vs. SCE and a redox potential of
(34*/4⋅−)=+1.35 V vs. SCE and a redox potential of  =+1.17 V vs. SCE (CH3CN) was determined for silyl enol ether 3 (see Supporting Information). We combined flavin 4 with model substrate 3 in a stoichiometric experiment (Figure 2) and irradiated the mixture with blue light in deuterated acetonitrile under an argon atmosphere. This resulted in clean conversion of the quinoid flavin to its reduced counterpart 5, concomitant to the formation of α,β-unsaturated ketone 6 in 94 % yield (determined versus NMR internal standard; see Supporting Information pages 38–40). When exposed to air, the solution quickly regained the typical flavin color, and oxidation of hydroquinone 5 to quinone 4 was observed. We concluded that photochemical desaturation is indeed feasible with molecular flavins and that all evidence points towards a single-electron oxidation pathway.21
=+1.17 V vs. SCE (CH3CN) was determined for silyl enol ether 3 (see Supporting Information). We combined flavin 4 with model substrate 3 in a stoichiometric experiment (Figure 2) and irradiated the mixture with blue light in deuterated acetonitrile under an argon atmosphere. This resulted in clean conversion of the quinoid flavin to its reduced counterpart 5, concomitant to the formation of α,β-unsaturated ketone 6 in 94 % yield (determined versus NMR internal standard; see Supporting Information pages 38–40). When exposed to air, the solution quickly regained the typical flavin color, and oxidation of hydroquinone 5 to quinone 4 was observed. We concluded that photochemical desaturation is indeed feasible with molecular flavins and that all evidence points towards a single-electron oxidation pathway.21

The stoichiometric Saegusa-Ito oxidation with a molecular flavin under inert conditions in a J Young NMR tube. Reaction conditions: Flavin 4 (1 equiv.) and silyl enol ether 3 (1 equiv.) were irradiated at λmax=451 nm in CD3CN solution for 2 h. The yield was determined versus NMR internal standard. No silylated reduced flavins were observed, which led to the conclusion that residual water serves as a source of protons (see Supporting Information).
In the next step, we aimed for achieving the Saegusa-Ito oxidation in a catalytic fashion (Table 1) with riboflavin tetraacetate (RFTA). We quickly realized that a basic additive (such as Na2HPO4) is required to avoid starting material hydrolysis to ketone 7. The key to success was finding a suitable sacrificial oxidant, which converts hydroquinoid flavin back to the quinoid form. Oxone® only facilitated approximately one turnover, while DDQ and K2S2O8 performed better with 34 % and 85 % yield, respectively (Entries 1–3). The oxidation with K2S2O8 releases hydrogensulfate,22 which is quenched by Na2HPO4. A stronger base such as Na3PO4 was not suitable, presumably as a result of catalyst decomposition (Entry 4).23 No reactivity was observed in the absence of flavin (Entry 5). We noticed that the reaction solution lost its typical yellow coloration after 2 h of irradiation, indicative of flavin catalyst decomposition. This prompted us to switch to modified flavin 4, and the reaction times could be extended to 8 h without risk of decoloration. Under these conditions, the unsaturated ketone 6 was formed almost quantitatively (Entry 6). No reactivity was observed without irradiation, and omitting the base additive diminished the yield (Entries 7 and 8).
|
||||||
Entry |
Flavin |
Oxidant |
Additive |
Time |
SMa |
Yield (6)a |
|---|---|---|---|---|---|---|
#1 |
RFTA |
Oxone® |
Na2HPO4 |
2 h |
85 % |
8 % |
#2 |
RFTA |
DDQ |
Na2HPO4 |
2 h |
n.d. |
34 % |
#3 |
RFTA |
K2S2O8 |
Na2HPO4 |
2 h |
n.d. |
85 % |
#4 |
RFTA |
K2S2O8 |
Na3PO4 |
2 h |
69 % |
n.d. |
#5 |
none |
K2S2O8 |
Na2HPO4 |
2 h |
>99 % |
n.d. |
#6 |
4 b |
K2S2O8 |
Na2HPO4 |
8 h |
n.d. |
>99 %b |
#7c |
4 b |
K2S2O8 |
Na2HPO4 |
8 h |
94 % |
n.d. |
#8 |
4 b |
K2S2O8 |
none |
8 h |
n.d. |
58 %d |

- [a] Determined versus NMR internal standard. [b]: Yield of isolated, volatile product 6: 74 %. [c]: No irradiation. [d]: Significant hydrolysis to ketone 7 (approx. 30 % yield). SM=Starting material. n.d.=none detected. RFTA=Riboflavin tetraacetate.
The scope of the catalytic desaturation was investigated with a series of silyl enol ethers (Figure 3). Our focus was on those substrates leading to aryl isopropenyl ketones, which are traditionally obtained by oxidation of the secondary alcohol or C−C bond formation reactions, while Saegusa-Ito protocols are not reported.24, 25 Alkyl-substituted arene substrates (products 8–10) and phenol ethers (products 11–13) led to clean conversion to the corresponding unsaturated ketones. Substrates with electron-deficient arene substituents were found to be less reactive, however, para-chloro (14) and para-fluoro substitution (15) worked reasonably well. Biphenyl derivative 16 and methyl ester 17 were equally successful. Our method is not limited to the formation of disubstituted olefins, and cyclohexene product 18 serves as an example of a trisubstituted analog. It is also not necessary to use aryl-substituted silyl enol ethers, which is highlighted by the clean oxidation to unsaturated ketone 19.

Substrate scope of the catalytic Saegusa-Ito reaction. Reaction conditions: See Table 1. Yields are determined versus NMR internal standard. [a] Yields of isolated material.
We then probed whether molecular oxygen can serve as an oxidant in our catalytic desaturation of silyl enol ether 3. Interestingly, a catalytic amount of flavin 4 also led to the formation of unsaturated ketone 6 under aerobic conditions, albeit in a poor yield of only 24 %. The reaction proceeded sluggishly, which can be explained by the competing reactivity of silyl enol ethers with singlet oxygen generated by flavin-mediated sensitization.13, 26 In line with this rationale, we identified silyloxy hydroperoxide 20 as a minor byproduct (Figure 4A).27 This reactive compound could be isolated, which prompted us to investigate its selective formation. We hypothesized that a less oxidizing flavin could be the key here since the excited state redox potential would not be sufficient to oxidize the silyl enol ether, but sensitization of molecular oxygen would still be possible. This succeeded with aminoflavin 21,28 which did not show any Saegusa-Ito oxidation activity under the standard conditions (see Table 1). However, when irradiated in the absence of an exogenous oxidant under an atmosphere of oxygen, silyloxy hydroperoxide 20 was formed in 62 % yield and could be fully characterized by NMR spectroscopy and HR-ESI-MS (see Supporting Information). This singlet oxygen ene reaction was also monitored by in situ NMR studies via illumination of the sample inside the magnet.29 This setup allowed us to follow and characterize the conversion to silyloxy hydroperoxide 20 and also to identify aromatic ketone 22 as a third product of this reaction (Figure 4B). The formation of both products can be rationalized based on the competition between a prototropic and a silatropic singlet oxygen ene reaction (Schenck ene reaction30).31

Probing molecular oxygen as a terminal oxidant. A: Products obtained from the reaction of silyl enol ether 3 under an oxygen atmosphere. B: In situ NMR monitoring of the reaction with substrate 3 (1 equiv.) in the presence of aminoflavin 21 (1 equiv.) in CD3CN. Irradiation was performed with a fiber optics setup and a 5 W LED (λmax=455 nm; see Supporting Information for details). [a] Yields determined versus NMR internal standard.
Encouraged by the stability of flavin catalyst 4 under the photochemical oxidation conditions, we next set out to investigate a sequential reaction that requires the same flavin catalyst to perform a second chemically distinct step in the same reaction vessel. In an orthogonal approach to the singlet oxygen generation under irradiation, reduced flavins are known to react with O2 via stepwise single-electron transfer leading to flavin hydroperoxides and, ultimately the release of hydrogen peroxide. All unsaturated ketones shown in Figure 3 are potential substrates for epoxidation reactions, which made us wonder whether the addition of a reductant to our reaction vessels after the Saegusa-Ito oxidation and exposure to O2 would trigger the one-pot conversion to epoxyketones. The latter are synthetically interesting reactive electrophiles32 and also serve as functional groups in proteasome inhibitors.33 In our one-pot reaction sequences (Table 2), we commenced with silyl enol ether 3 and used the catalytic conditions (c.f., Table 1) that resulted in the nearly quantitative formation of the α,β-unsaturated ketones. Our focus was initially on identifying a suitable reductant for the second reaction step. Zinc is often used as a reductant for flavins,34 yet its application in our reaction sequence resulted only in trace product formation (Entry 1). A slight improvement was found with Hantzsch ester (HEH) in combination with a carbonate base and methanol as a co-solvent (Entries 2 and 3). Switching to Cs2CO3 and adding a small amount of water (0.1 equiv.) improved the yield to 25 %, presumably by increasing the carbonate solubility in the organic solvent mixture (Entry 4). Increasing the reaction temperature to 40 °C led to faster epoxidation and increased levels of product formation (Entries 5 and 6). All yields of epoxyketone 23 refer to the two-step reaction sequence and are based on the silyl enol ether starting material.
|
||||
Entry |
Reductant |
Base |
Co-Solvent |
Yielda |
|---|---|---|---|---|
#1 |
Zinc (5 equiv.) |
None |
Water (10 % v/v) |
2 % |
#2 |
HEH (4 equiv.) |
None |
MeOH (25 % v/v) |
n.d. |
#3 |
HEH (2 equiv.) |
K2CO3 |
MeOH (25 % v/v) |
4 % |
#4 |
HEH (2 equiv.) |
Cs2CO3 |
MeOH (25 % v/v)b |
25 % |
#5 |
HEH (2 equiv.) |
Cs2CO3 |
MeOH (25 % v/v)c |
53 % |
#6 |
HEH (2 equiv.) |
Cs2CO3 |
MeOH (25 % v/v)b,c |
60 %d |
- All sequential catalytic reactions were initially irradiated for 8 h (Saegusa-Ito oxidation) and then stirred overnight with reductant under an atmosphere of oxygen. [a] Determined versus NMR internal standard. [b] With 0.1 equiv. water. [c] Reaction performed at 40 °C. [d] Yield of isolated material: 46 %.
These results prompted us to investigate whether analogous sequential reaction conditions can also be applied to the entire set of Saegusa-Ito substrates shown in Figure 3. We were pleased to see that all unsaturated ketones were also smoothly converted into the corresponding epoxyketones (Figure 5), with the only exception of ester 17, which resisted oxygenation. Alkyl-substituted arenes were tolerated best (24–26), while electron-rich arenes showed slightly lower levels of product formation (27–29). Halide- and aryl-substitution of the aromatic ketones was well tolerated and led to epoxyketones 30 to 32. Even the trisubstituted epoxide 33 and aliphatic derivative 34 were obtained, albeit in lower yields. In the latter cases, conversion was observed to be incomplete and much slower.

Products of the one-pot desaturation epoxidation sequence. Reaction conditions: See Table 2. Yields were determined versus NMR internal standard [a] Yields of isolated material.
Sequential flavin catalysis is not limited to the combination of desaturation and epoxidation. Subsequent to the Saegusa-Ito oxidation step, flavin-catalysis also successfully generates CF3-radicals from Langlois reagent (NaSO2CF3),35 which results in addition product 35 in 60 % yield over two steps (Figure 6A). In an orthogonal approach, flavin catalysis leads to C−C bond formation by radical addition to benzylidenemalononitrile (Figure 6B). In analogy to the initial report of this reactivity by Ooi,21a this requires an allylic radical which is generated by deprotonation of the silyl enol ether radical cation after initial flavin-mediated oxidation of substrate 36. While dichloromethane performed poorly as a solvent in the Saegusa-Ito reaction (see Supporting Information page 42), it was suitable here. The anionic flavin semiquinone radical is proposed to deprotonate the substrate intermediate, but adding the additional base Na2HPO4 led to further improvement (63 % yield without base; see Supporting Information page 89). The immediate silyl enol ether product 37 can then be subjected to a subsequent flavin-mediated Saegusa-Ito oxidation, which leads to α,β-unsaturated ketone 38 (individual steps: 67 % and 71 % yield; one-pot, two-step procedure: 48 % yield). An aerobic halogenation of phenolic substrates with LiBr28 is also possible after Saegusa-Ito oxidation (see Supporting Information pages 85–87).

Examples of the application of sequential flavin catalysis: Saegusa-Ito oxidation and subsequent radical trifluoromethylation leads to ketone 35 (A). The β-functionalization of silyl enol ether 36 prior to Saegusa-Ito oxidation results in elongated, α,β-unsaturated ketone 38 (B). Yields were determined versus NMR internal standard.
In summary, we report an unprecedented one-pot strategy for directly converting silyl enol ethers into α,β-epoxyketones which relies on the combination of two orthogonal flavin catalyst activities, namely the photochemical single-electron oxidation and the reductive activation of O2. While common for flavoenzymes, our two-step reaction sequence with a single molecular flavin catalyst paves the way for similar combination strategies to be used in the organic laboratory. Synthetically valuable sequential reactions are anticipated, for example, by connecting the reductive and oxidative activity of flavins with their function as organocatalysts.
Acknowledgments
The Fonds der Chemischen Industrie (FCI, Ph.D. Fellowship to A.W and Liebig Fellowship to G.S) is gratefully acknowledged. The project was funded by the Deutsche Forschungsgemeinschaft (Emmy Noether Programme, STO 1175/3-1). We thank J. Großkopf for spectroscopic measurements and M. Schick for HPLC separations. Our group is supported by the Technical University of Munich through the Junior Fellow Programme. G.S. is very grateful to Prof. T. Bach for his continuous support. Open Access funding enabled and organized by Projekt DEAL.
Conflict of interest
The authors declare no conflict of interest.
Open Research
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.

