

Controlling Metal-Oxide Reducibility for Efficient C−H Bond Activation in Hydrocarbons
Graphical Abstract
Through catalyst post-treatment in an NH3 ⋅ H2O solution, structural and chemical properties of supported metal oxide species can be controlled because of dissolution-recrystallization of amorphous or/and low-crystalline support in the parent catalyst. The method is applicable for tailoring the catalyst reducibility and thus its activity for C−H activation in hydrocarbons.

Abstract
Knowing the structure of catalytically active species/phases and providing methods for their purposeful generation are two prerequisites for the design of catalysts with desired performance. Herein, we introduce a simple method for precise preparation of supported/bulk catalysts. It utilizes the ability of metal oxides to dissolve and to simultaneously precipitate during their treatment in an aqueous ammonia solution. Applying this method for a conventional VOx−Al2O3 catalyst, the concentration of coordinatively unsaturated Al sites was tuned simply by changing the pH value of the solution. These sites affect the strength of V−O−Al bonds of isolated VOx species and thus the reducibility of the latter. This method is also applicable for controlling the reducibility of bulk catalysts as demonstrated for a CeO2−ZrO2−Al2O3 system. The application potential of the developed catalysts was confirmed in the oxidative dehydrogenation of ethylbenzene to styrene with CO2 and in the non-oxidative propane dehydrogenation to propene. Our approach is extendable to the preparation of any metal oxide catalysts dissolvable in an ammonia solution.
Introduction
Providing the fundamentals for rational design and synthesis of heterogeneous catalysts with a certain kind of supported species or/and bulk structure(s), that are the key descriptors of catalyst activity, product selectivity and on-stream stability, is an insistently pursued task towards sustainable development. The precise synthesis of solid catalysts is, however, challenging due to the heterogeneity of crystal structure(s), crystallinity, and/or chemical composition/distribution from nano to atomic/molecular scales. Moreover, there are great difficulties in identifying of catalytically active sites and/or the mechanism of their formation associated with surface heterogeneity.1 Indeed, a lot of efforts ranging from first-principle calculations to experimental investigations have been devoted to the development of versatile strategies for purposeful preparation of efficient heterogeneous catalysts.2 For example, top-down/bottom-up assembly,3 atomic layer deposition,4 flame spray pyrolysis,5 solution combustion synthesis,6 and plasma-assisted routes7 have been applied. Although these strategies have provided important fundamentals for preparation of well-defined structures/phases, their application for large-scale catalyst preparation is commonly failed to meet the economic and/or technological criteria in comparison with the industrially relevant approaches including impregnation, precipitation, and hydrothermal or sol-gel syntheses. The latter methods cannot, however, ensure the creation of controlled structures, resulting in inhomogeneous catalyst structure(s) and composition(s).
Catalysts based on oxides of vanadium, chromium, aluminum, cerium, molybdenum, or zinc are widely used in different industrial reactions involving activation of C−H bonds in various hydrocarbons.8 As their preparation typically involves a precipitation process, the inherently unmanageable precipitation/recrystallization rates of hydrous oxides or carbonates in aqueous solutions result in the heterogeneity in structure, crystallinity, and local composition of metal oxide catalysts. In this case, great efforts are practiced to regulate the precipitation rates through adding complex agents,9 in addition to a precise control of synthesis parameters such as temperature and pH value. However, the desired structural control during the large-scale production of solid catalysts is still a great issue. This can be exemplarily reflected by widely used γ-alumina, the structure, crystallinity, and textural properties of which are always varied to different extents from batch to batch of the same producer or individual manufacturers.10 In fact, each precipitation process in an aqueous solution is based on the precipitation-dissolution dynamic equilibrium of metal cations as established and documented in the textbook of heterogeneous catalysis.11 Thus, if a given metal oxide is added to an aqueous solution, its dissolution and re-precipitation may also occur dynamically.12
Herein we introduce a simple method with a potential of large-scale applications to tailor the structure of metal-oxide catalysts and to efficiently improve their performance in dehydrogenation reactions of different hydrocarbons. Our working hypothesis was that structural and chemical properties of supported metal oxide species can be controlled through catalyst post-treatment in an NH3 ⋅ H2O solution because of dissolution-recrystallization of amorphous or/and low-crystalline supports in the parent catalysts. This methodology was validated on the examples of VOx−Al2O3 and CeO2−ZrO2−Al2O3 materials tested in the oxidative dehydrogenation of ethylbenzene to styrene with carbon dioxide (CO2−ODEB) and the non-oxidative propane dehydrogenation to propene (PDH). The activity of isolated VOx species depends on the strength of V−O−Al bond, which is determined by the content of coordinatively unsaturation of aluminum cations on the support surface. The concentration of such defective sites can be controlled through our preparation method. Redox properties of ceria can also be adjusted and are relevant for the dehydrogenation activity.
Results and Discussion
A VOx−Al2O3 catalyst with a V2O5 loading of 3 wt % (abbreviated as s-3VA) synthesized by a sol-gel method13 is introduced to demonstrate our approach schematically shown in Figure 1a. The post-treatment of s-3VA in an NH3 ⋅ H2O solution with a pH of 13 led to the post-treated catalyst s-3VA-pH 13. As revealed by transmission electron microscopy (TEM), high resolution TEM (HRTEM) and high-angle annular dark field scanning TEM (HAADF-STEM), s-3VA is composed of agglomerated nanosheets (Figure 1b and Figure S1a,b) and Al2O3 particles (5–10 nm) with a polycrystalline structure (Figure 1c and Figure S2a,b). The lattice fringes of (400) and (131) with a characteristic acute angle of 72.5° for γ-Al2O3 were identified (Figure 1d). In contrast, s-3VA-pH 13 consists of larger particles of Al2O3 with relatively regular edges and morphologies (Figure 1e and Figure S1c,d). The crystallinity of γ-Al2O3 is obviously improved after the post-treatment; the diffraction spots of FFTs are much sharper and brighter than those of s-3VA (Figure 1f,g and Figure S2c,d). No crystalline V2O5 was detected both in s-3VA and s-3VA-pH 13 (Figure 1c,f and Figure S2a,c). Vanadium is well distributed on the surface of catalysts as revealed by the elemental mapping of energy dispersive X-ray spectroscopy (EDX, Figure 1h,i).

a) Illustrative procedure for the post-treatment of the s-3VA catalyst in an NH3 ⋅ H2O solution. b–i) images of TEM, HRTEM, and EDX-elemental mapping for s-3VA (b, c, d and h) and s-3VA-pH 13 (e, f, g and i).
To find the reason behind the above-discussed structural modifications of Al2O3, s-3VA was additionally treated in an NH3 ⋅ H2O solution with pH of 9 or 11 to yield the s-3VA-pH 9 and s-3VA-pH 11 catalysts. Importantly, inductively coupled plasma mass spectrometry (ICP-MS) measurements confirmed the presence of Al3+ ions in the supernatant after post-treatment of s-3VA. The concentration of aluminum increased from 6.6 to 32.7 mg/L with an increase in the pH value of NH3 ⋅ H2O solution from 9 to 13 (Table S1). X-ray diffraction (XRD) analysis of post-treated s-3VA after drying (not calcined) indicated the presence of hydrous Al2O3 species (Figure 2a) mainly in the form of pseudoboehmite (PDF#83-2384) in s-3VA-pH 9-P, gibbsite (PDF#33-0018) in s-3VA-pH 11-P, and nordstrandite (PDF#24-0006) in s-3VA-pH 13-P.14 Thus, the dissolution and the re-precipitation of Al2O3 took place during the s-3VA post-treatment.

a) XRD patterns and b) the weight loss during TG analysis of post-treated VOx−Al2O3 precursors. c) XRD patterns, d) the content of Alcus, e) in situ UV Raman spectra and f) the Eg values for the parent and post-treated VOx−Al2O3 catalysts. g) A schematic showing the dissolution-precipitation process of the catalyst precursor in the NH3 ⋅ H2O solution and its effects on the crystallization of Al2O3 and the strength of VOx−Al2O3 interactions. Symbols #, o, * and+in the XRD patterns stand for the pseudoboehmite (PDF#83-2384), gibbsite (PDF#33-0018), nordstrandite (PDF#24-0006), and γ-Al2O3 (PDF#50-0741), respectively.
As revealed by thermal gravimetric (TG) and differential scanning calorimetry (DSC) analyses (Figure 2b and Figure S3a,b), the amount of hydrous Al2O3 increased in the order of s-3VA-pH 9-P<s-3VA-pH 11-P<s-3VA-pH 13-P. This indicates that higher concentration of OH− can accelerate dissolution-precipitation processes, leading to the formation of higher amounts of hydrous Al2O3. This compound is known to transform into γ-Al2O3 via dehydroxylation/dehydration during calcination at 550 °C.15 Only diffractions characteristic of γ-Al2O3 (PDF#50-0741) were observed in the XRD patterns of the parent and post-treated (calcined) samples (Figure 2c), coinciding with HRTEM observations (Figure 1c,f and Figure S2a,c). The intensity of the main diffractions increased in the order of s-3VA<s-3VA-pH 9<s-3VA-pH 11<s-3VA-pH 13, while the full width at half maximum (FWHM) of the (440) diffraction decreased from 4.5° to 1.8° (Figure S4). These changes indicate the recrystallization of amorphous and low-crystalline Al2O3 in the parent s-3VA catalyst during the post-treatment and confirm the importance of the solution pH value for the crystallinity of Al2O3.
To check if the structure of hydrous Al2O3 affects its transformation into crystalline γ-Al2O3, a VOx−Al2O3 catalyst (3 wt % V2O5, abbreviated as e-3VA) with an amorphous structure (Figure S5a) was on purpose synthesized by an evaporation-induced self-assembly method.16 It was also subjected to a post-treatment in an NH3 ⋅ H2O solution with pH of 13. In contrast to s-3VA-pH 13-P with the dominant nordstrandite phase (Figure 2a), both the nordstandite and pseudoboehmite phases were formed in the treated but non-calcined e-3VA-pH 13-P (Figure S5a). The amount of hydrous Al2O3 determined by TG analysis in this sample is very close to that in s-3VA-pH 13-P (Figure S5b). However, s-3VA-pH 13 possesses a clearly higher amount of crystalline γ-Al2O3 than e-3VA-pH 13 after these samples were calcined at 550 °C (Figure S5c). Thus, comparing with pseudoboehmite, the dehydroxylation/dehydration of nordstandite led to a higher crystallinity of γ-Al2O3. This is consistent with the higher crystallinity of γ-Al2O3 in s-3VA-pH 13 than that in s-3VA-pH 9, because the nordstrandite and pseudoboehmite phases are dominant in s-3VA-pH 13-P and s-3VA-pH 9-P, respectively. Thus, both the amount and the structure of hydrous Al2O3, which are determined by the pH value of NH3 ⋅ H2O solution during the post-treatment, affect the crystallinity of γ-Al2O3 in the post-treated catalysts.
27Al magic-angle-spinning (MAS) NMR measurements were additionally carried out to analyze the chemical environment of Al species in the parent and post-treated samples. Three peaks at about 6, 31 and 70 ppm are present in the obtained spectra (Figure S6a), which can be assigned to Al3+ species in octahedral (AlO6), pentahedral (AlO5) and tetrahedral (AlO4) coordination, respectively.17 The concentration of coordinatively unsaturated Al sites with the AlO5 coordination (Alcus) decreases from 19.1 % to 9.3 % in the order s-3VA>s-3VA-pH 9>s-3VA-pH 11>s-3VA-pH 13 (Figure 2d), while the concentration of coordinatively saturated Al sites (Alcs) with the AlO6 or AlO4 coordination shows an opposite order (Figure S6b), which can be attributed to the increased crystallinity of γ-Al2O3.
Although a small quantity of vanadium ions was detected by ICP-MS in the supernatant after post-treatment of s-3VA (Table S1), no crystalline V2O5 was formed in the post-treated catalysts (Figure 1f and Figure S2c). This suggests that no recrystallization process related to VOx occurred during the catalyst treatment. To find out the reason behind this observation, a controlled dissolution experiment was performed. The crystalline V2O5 sample was completely dissolved in the NH3 ⋅ H2O solution within 0.5 h (Figure S7). This indicates that the dissolution-precipitation processes, which take place on the surface of amorphous and low-crystalline Al2O3 in the parent s-3VA catalyst during the post-treatment, do not occur in the case of crystalline V2O5. Thus, any recrystallization of VOx species can be ruled out.
The content of vanadium in the parent and post-treated VOx−Al2O3 determined by ICP-MS was same within the experimental error (1.55±0.05 wt %, Table S2). A symmetric V2p3/2 peak with the binding energy of 517.6 eV and FWHM of 1.8±0.1 eV assigned to V5+ was observed in the X-ray photoelectron spectra (XPS) of all catalysts (Figure S8). Thus, the post-treatment did not affect the content and the oxidation state of vanadium.
The in situ UV Raman spectrum of the parent s-3VA catalyst contains only two distinct shifts at 901 and 1015 cm−1 (Figure 2e), which are characteristic for the vibrations of V−O−Al and V=O bonds in monomeric VOx species, respectively.18 The high dispersion of vanadium is due to its low surface density of 0.44 V atoms/nm2 (Table S2), which is far below the theoretical monolayer density of 2.3 V atoms/nm2 for monomeric VO4.19 Although no additional bands are present in the spectra of the post-treated catalysts, the Raman shift due to the vibration of V−O−Al bonds decreases from 901 to 886 cm−1 in the order s-3VA>s-3VA-pH 9>s-3VA-pH 11>s-3VA-pH 13. The red shift of this characteristic peak was also observed in the spectrum of VOx/γ-Al2O3 after the introduction of sulfur.20 It is commonly explained by weakening the strength of the bonds as reported in previous studies.21 Further understanding on this phenomenon can be obtained from in situ UV/Vis spectra of the catalysts under anhydrous conditions (Figure S9). The therefrom calculated edge energy (Eg) of VOx decreases from 3.75 (s-3VA) to 3.62 (s-3VA-pH 9), 3.53 (s-3VA-pH 11), and 3.50 eV (s-3VA-pH 13) (Figure 2f). Generally, Eg depends on the polymerization degree of VOx and cationic ligand of the support.22 As only monomeric VO4 species are present in the parent and post-treated catalysts, the change in the Eg values may originate from the coordination degreed of Al sites in the support surface, which are connected with the VOx species. Among these four catalysts, the parent s-3VA with amorphous and low-crystalline Al2O3 shows the highest Eg values, indicating the strongest VOx−Al2O3 interaction. This coincides with the highest strength of V−O−Al bonds as proved by the Raman results (Figure 2e). The strength and the Eg value decrease with an increase in the pH value of NH3 ⋅ H2O solution used for catalyst post-treatment. These facts can be ascribed as the decreased amount of Al sites strongly interacting with VOx species. In this case, Alcus typically characterizing the strong interactions with VOx species should be such sites, which is also supported by the fact that Alcus content actually decreases with an increase in the crystallinity of γ-Al2O3 (Figure 2d and Figure S6a,b).
Regardless of the pH value of the post-treatment solution, the amount of H2 consumed during temperature-programmed reduction tests with H2 (H2-TPR) is same for all catalysts, which corresponds to the reduction of V5+ to V4+ (+4.15±0.05, Table S3).18b, 23 This can be attributed to the exclusively monomeric VOx species over these catalysts as revealed by Raman results (Figure 2e). However, the temperature of the maximal rate of H2 consumption decreases from 520 °C for the parent s-3VA to 514–504 °C for the post-treated s-3VA with an increase in the pH value of the NH3 ⋅ H2O solution (Figure S10). This indicates that the reducibility of monomeric VOx increases in the order of s-3VA<s-3VA-pH 9<s-3VA-pH 11<s-3VA-pH 13. Considering that monomeric VOx species only contains V−O−Al and V=O bonds, such improvement can be explained by weakened interactions between VOx and Al2O3 as indicated by the decreased Eg value and strength of V−O−Al bonds. Summarizing these results and their discussion, the dissolution-precipitation mechanism and the induced effect of the post-treatment on VOx−Al2O3 catalysts were developed, which are schematically shown in Figure 2g.
We also proved the potential of the developed method for preparation of bulk catalysts. A representative CeO2, ZrO2 and Al2O3 mixed oxide (28 wt % CeO2−ZrO2 with a Ce/Zr molar ratio of 6/4, abbreviated as s-28CZA) was synthesized through a sol-gel method and was post-treated in an NH3 ⋅ H2O solution with a pH of 13 (abbreviated as s-28CZA-pH 13). Amorphous Al2O3, CeO2 and ZrO2 were identified in s-28CZA (Figure 3a and Figure S11a), which may be explained as that Al2O3 hinders the formation of crystalline CeO2−ZrO2 solid solutions.24 Importantly, the post-treated s-28CZA-pH 13 is composed of well-crystallized γ-Al2O3 and cubic Ce-based oxide with oxygen defects (Figure 3a and Figure S11a). To identify the phase properties, pure CeO2 (abbreviated as s-Ce) and Ce0.6Zr0.4O2 solid solution (abbreviated as s-CZ) also synthesized via the sol-gel method were characterized by visible Raman spectroscopy (Figure S11a) and XRD (Figure S12). In comparison with the XRD pattern of s-Ce, the addition of Zr to CeO2 (s-CZ) led to a shift of the CeO2-related reflections to larger 2θ values (Figure 3b, Figure S12) accompanying with decreased lattice parameter (Table S4). This is due to the formation of Ce0.6Zr0.4O2 solid solution via the insertion of Zr4+ into the CeO2 lattice.25 Noteworthy, the (111) diffraction and the lattice parameter of s-28CZA-pH 13 are same as those of s-CZ (Figure 3b and Table S4), suggesting the same composition of the solid solution in these two catalysts.24a, 26 XRD results of dried precursor, i.e., s-28CZA-pH 13-P clearly show the presence of hydrous CeO2, nordstrandite and gibbsite (Figure S13a), indicating that the dissolution and the precipitation of amorphous and/or low-crystalline Al2O3 and CeO2 took place simultaneously during the post-treatment of the parent s-28CZA material. Although the hydrous ZrO2 was not detected by XRD perhaps because of its amorphous nature (Figure S13b), its formation is expected if the solubility product constants of hydrous ZrO2 (pKsp=48.2) and hydrous CeO2 (pKsp=47.7) are taken into account. The s-28CZA-pH 13 sample shows a clearly higher reducibility (Figure S14) than the parent s-28CZA. Moreover, in comparison with s-CZ, s-28CZA-pH 13 has a higher concentration of oxygen defects as confirmed by the Raman and Ce 3d XPS patterns (Figures S11b and S15a,b). This is due to smaller crystal size of Ce0.6Zr0.4O2 in s-28CZA-pH 13 (5.2 nm) than that in s-CZ (7.8 nm, Table S4).

a) Full XRD patterns of s-28CZA and s-28CZA-pH 13. b) Enlarged (111) reflections of the cubic phase over s-Ce, s-CZ and s-28CZA-pH 13 (+: γ-Al2O3 (PDF#50-0741); ◊: Ce0.6Zr0.4O2 solid solution (PDF#38-1439)).
The oxidative dehydrogenation of C2-C4 alkanes or ethylbenzene (EB) with carbon dioxide is an attractive technology for the efficient production of olefins or styrene in tandem with the CO2 reduction to CO.8a, 8b V- or Ce-based oxides are promising catalysts for CO2−ODEB, and their activity is closely connected with their structural and chemical properties.18b, 25 Now we demonstrate the potential of the developed catalysts for the CO2−ODEB reaction. In agreement with our previous findings,18b, 27 both the parent and post-treated pure Al2O3 and ZrO2(28 wt %)-Al2O3 oxides are essentially inactive (Figure S16). Thus, the below discussion can be safely focused on the role of supported VOx species or Ce-based phases in this reaction.
The apparent turnover frequency (TOFEB) over V- and Ce-based oxides is defined and calculated as the number of EB molecules converted per V or Ce atom per hour. The TOFEB value of VOx−Al2O3 catalysts increases from 22.8 to 41.0 h−1 in the order s-3VA<s-3VA-pH 9<s-3VA-pH 11<s-3VA-pH 13 (Figure 4a), while the selectivity to styrene is above 99.0 % for all catalysts (Figure S17). The enhanced activity of post-treated catalysts can be reasonably associated with the weakened interaction between VOx and Al2O3 because the oxidation state of vanadium (V5+), the molecular structure of VOx species (exclusively monomeric VOx), and the amount of reducible VOx species are essentially same for all these catalysts.

a) TOFEB of the parent and post-treated VOx−Al2O3 catalysts in CO2-ODEB. b) Time-on-stream stability of s-3VA-pH 13 and s-28CZA-pH 13. c) STC (space-time conversion of EB) versus Rd (relative deactivation rate in CO2-ODEB) for the catalysts developed in this work and reported in literatures (The number from 1 to 16 represents the catalyst of the corresponding entry in Table S5, respectively). d) Time evolved in situ UV/Vis spectra, and e) ΔF(R) at 700 nm during the reduction of the fully oxidized VOx species over s-3VA by H2 (Open circles: experimental data, solid line: simulated results). f) TOFEB over the parent and post-treated VOx−Al2O3 catalysts as a function of k′(H2) calculated by fitting ΔF(R) at 700 nm.
The TOFEB value of the s-28CZA-pH 13 catalyst is about 13 times higher than that of s-28CZA (10.6 vs. 0.8 h−1, Figure S18). This result together with the catalyst characterization data (Figure 3a and Figure S11a) proves that amorphous CeO2 in s-28CZA is almost inert for activating C−H bonds in EB, while the crystalline Ce0.6Zr0.4O2 solid solution in s-28CZA-pH 13 is highly active. A similar conclusion about the effect of crystallinity was also reported for pure ZrO2-catalyzed PDH reaction.28 Thus, coordinatively unsaturated metal cations (Mecus) in crystalline CeO2 or ZrO2 are required to break C−H bonds in EB or C3H8 molecules. This is supported by a higher TOFEB value of s-28CZA-pH 13 than s-CZ (Figure S18) due to the higher concentration of oxygen defects and Ce3+ (direct indicators for Mecus, Figures S11b and S15a,b) and higher reducibility of Ce0.6Zr0.4O2 in s-28CZA-pH 13 (Figure S14).
The developed s-3VA-pH 13 and s-28CZA-pH 13 catalysts also show high long-term stability. No obvious deactivation was observed during 45 h on EB stream under industrially relevant conditions (Figure 4b and Figure S19a,b). We also benchmarked our catalysts against the state-of-the-art V- or Ce-based catalysts used in the CO2−ODEB reaction at 550 °C and comparable contact times (Table S5). For this purpose, the space-time conversion of EB (STC) defined as the molar amount of EB converted per mole of V or Ce per hour and relative deactivation rate (Rd) expressed as the ratio of (STCinitial−STCfinal)/STCinitial×100 % (STCinitial and STCfinal stand for STC at the beginning and at the end of the reaction, respectively) were calculated. The developed s-3VA-pH 13 and s-28CZA-pH 13 show the highest initial STC and the lowest Rd among their previously reported counterparts (Figure 4c). Moreover, the high stability of the developed catalysts is further confirmed by the deactivation rate constant (kd, Table S6) calculated on the basis of a first-order deactivation model.29
Time-resolved in situ UV/Vis spectroscopy was applied to check if redox kinetics of supported VOx species and bulk CeO2 can be a descriptor in interpreting their catalytic activity for CO2−ODEB. Following previously reported studies,30 H2 was used as a reducing agent instead of EB to exclude any potential effect of carbon-containing deposits on reaction-induced changes in the UV/Vis spectra. Figure 4d exemplarily shows the spectra (expressed as the Kubelka–Munk function, F(R)) of fully oxidized s-3VA and after different times on H2 stream (10 %vol H2/N2) at 550 °C. The former spectrum is characterized by the oxygen ligand to metal charge transfer (LMCT) bands of V5+ with the absorption maxima below 400 nm.31 The intensity decreases with rising time on H2 stream due to the reduction of V5+ to V4+/V3+. The presence of the latter species is supported by the appearance of absorption bands in the range of 600–800 nm, which are assigned to d-d transitions of V4+/V3+.30a Their intensity remained unchanged after about 10 min in H2 due to reaching a steady-state concentration of such VOx species. Similar changes were also observed in the spectra of Ce-based catalysts (Figure S20).
 (1)
(1)where  is the concentration of reduced VOx species,
is the concentration of reduced VOx species, 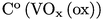 is the total concentration of oxidized VOx species, which participate in the reduction.
is the total concentration of oxidized VOx species, which participate in the reduction.
The simulated responses and the experimental ΔF(R) at 700 nm for s-3VA are presented in Figure 4e, while such data for s-3VA-pH 9, s-3VA-pH 11 and s-3VA-pH 13 are given in Figure S21a–c. The model in equation 1 correctly describes the experimental data for all catalysts. The derived k′(H2) values of s-3VA, s-3VA-pH 9, s-3VA-pH 11 and s-3VA-pH 13 are 0.66, 0.75, 0.83 and 0.96 min−1, respectively. There exists a positive correlation between the TOFEB and k′(H2) as seen in Figure 4f. Thus, the post-treatment of the catalyst in the NH3 ⋅ H2O solution accelerates the kinetics for the reduction of oxidized VOx species, which is beneficial for the conversion of EB to styrene. A similar conclusion was also established for s-CZ versus s-28CZA-pH 13 (Figures S18 and S22a,b).
Supported VOx catalysts are also promising candidates for the non-oxidative dehydrogenation of lower alkanes such as propane.8b, 32 V4+/V3+Ox species are considered to be the active sites in the PDH reaction.33 To check if their intrinsic activity can be regulated through altering the V−O-support interactions, the developed V-containing catalysts were tested in the PDH reaction at 600 °C using an industrially relevant feed consisting of 40 vol %C3H8 in N2. The catalysts were initially reduced in a flow of 50 vol % H2/N2 to remove active lattice oxygen species and thus to exclude the oxidative dehydrogenation of propane during a few first minutes on stream.33c Moreover, coke deposition over VOx catalysts for PDH is resisted by V−OH species formed during the H2 pre-treatment.34 As indicated by the results in Figure 5a, the apparent TOFpropane increases in the order s-3VA<s-3VA-pH 9<s-3VA-pH 11<s-3VA-pH 13 while the selectivity to propene is about 95.0 % for all catalysts (Figure S23). Since the molecular structure of exclusively monomeric VOx is essentially same in all catalysts, the enhanced activity of post-treated catalysts should be attributed to weakened interactions between VOx and Al2O3 as suggested in references.33a, 35 For the catalysts with too weak interactions between active sites (e.g., Pt) and support, restructuring may occur during the reaction at high temperatures, leading to the loss of activity.16a, 17a, 36 Thus, an additional test consisting of 12 PDH/oxidative regeneration cycles was performed over s-3VA-pH 13. No obvious loss in the propane conversion and propene selectivity could be identified from cycle to cycle (Figure 5b). Thus, VOx species are not restructured even under severe PDH conditions, which is consistent with the excellent durability of the impregnated VOx/γ-Al2O3 catalyst for the PDH reaction as reported in a very recent study.37 Moreover, as indicated from the apparent TOFpropane of s-28CZA and s-28CZA-pH 13 (4.2 versus 8.4 h−1, Figure S24), the post-treatment of s-28CZA also improves its activity for the PDH reaction.

a) TOFpropane over the parent and post-treated VOx−Al2O3 catalysts. b) Time-on-stream propane conversion and propene selectivity of PDH over the s-3VA-pH 13 catalyst regenerated for 12 cycles. For each cycle, the temperature was kept constantly at 600 °C, and it contained three stages, i.e., a reduction stage in a 50 vol % H2/N2 mixture for 30 min, a PDH reaction stage fed with 40 vol % C3H8/N2 for 2 h, and a regeneration stage in the air for 30 min. c,d) The height-normalized (the X axis was transformed into a dimensionless form according to the reference38) transient responses of C3H8 (green), C3H6 (black) and H2 (red) after pulsing a C3H8/Ar=1/1 mixture (c) and D2 (blue) and HD (cyan) after pulsing a H2/D2/Ar=1/1/1 mixture (d) over s-3VA-pH 13 reduced at 600 °C.
To derive fundamental insights into the PDH reaction over s-3VA, s-3VA-pH 9, s-3VA-pH 11 and s-3VA-pH 13, we carried out pulse experiments with C3H8 or H2/D2 in the temporal analysis of products (TAP) reactor at 600 °C in high vacuum. The catalysts were reductively treated to mimic the reduction state of vanadium as in the steady-state PDH tests. Propene and hydrogen were detected after pulsing of a C3H8/Ar=1/1 mixture (Figure 5c and Figure S25a–c). The catalytic activity in terms of the propene formation can be ordered as s-3VA<s-3VA-pH 9<s-3VA-pH 11<s-3VA-pH 13 (Figure S25d), which is same to that of the steady-state PDH tests (Figure 5a). This further confirms the improved activity of the post-treated catalysts for activating C−H bond in propane.
The time scale of gas-phase components in Figure 5c and Figure S25a–c was converted into a dimensionless form as suggested in the reference.38 The details can be found in the Supporting Information. Such transformation is required for correct comparing the order of appearance of C3H8, C3H6 and H2 since their diffusion velocity is strongly different. As expected, the responses of C3H6 and H2 as products appear after the response of the C3H8 reactant. However, the maximum of the dimensionless time of the C3H6 response is obviously shorter than that of the H2 response (Figure 5c and Figure S25a–c). This indicates that the rates for the formation of C3H6 and H2 are not equal under transient conditions, which is different from the steady-state observations. It is commonly accepted that the PDH reaction occurs via the consecutive activation of two C−H bonds in one propane molecule, leading to the formation of adsorbed C3H6 and H2 on the catalyst surface. After desorption, gaseous C3H6 and H2 are released. Thus, based on the position of the maxima of the C3H6 and H2 responses, it can be proposed that the rate-limiting step in the VOx-catalyzed PDH reaction is the H2 formation but not the cleavage of C−H bonds in C3H8. This statement is further supported by the results of temperature-programmed surface reaction of propane (TPSR-C3H8) over s-3VA-pH 13 (Figure S26). The staring temperature of H2 appearance in the gas phase (350 °C) is clearly higher than that of C3H6 (300 °C).
To check if the delayed release of H2 in the C3H8 pulse tests could be caused by its potential reactions with surface OH groups, hydrogen isotopic exchange experiments were carried out using a H2/D2/Ar=1/1/1 mixture. As HD was observed in outlet gases (Figure 5d and Figure S27a–c), the breaking of D−D bonds in D2 molecules and the formation of H−D bonds happened over all of the catalysts. Moreover, the order of catalyst activity for the H/D exchange reaction (Figure S27d) is same to that for the PDH reaction over the investigated catalysts (Figure S25d). Irrespective of the catalysts, the dimensionless time for releasing gaseous HD (Figure 5d and Figure S27a–c) is much shorter than that for the appearance of gaseous H2 (Figure 5c and Figure S25a–c). This clearly indicates that the delayed appearance of H2 in comparison with C3H6 cannot originate from the secondary transformation and desorption of H2 in the course of the PDH reaction. Thus, the formation of H2 after the activation of C−H bonds in propane is reasonably proposed as the rate-determining step in the VOx catalyzed PDH reaction. Considering this conclusion and the PDH activity in Figure 5a, we put forward that the strength of V−O−Al bond affects the ability of reduced isolated VOx species to form gas-phase H2 through recombination of two surface H species originated upon the breaking of C−H bonds in propane.
Conclusion
In summary, we have demonstrated that a simple post-treatment of VOx−Al2O3 and CeO2−ZrO2−Al2O3 in an aqueous NH3 solution is an industrially applicable method to tailor structural and redox properties of these catalysts, which are determined by dissolution/precipitation/recrystallization processes of amorphous or less crystalline metal oxides in the treatment solution with varied pH values. The accordingly developed VOx-containing or Ce-based catalysts outperform the corresponding state-of-the-art catalysts in terms of their activity in the CO2−ODEB reaction. For VOx−Al2O3 catalysts, such treatment enables control of the concentration of Alcus sites, which determine the reducibility of isolated V5+ sites. This catalyst property is a key descriptor affecting the rate of styrene formation as well as the rate of the PDH reaction. The reducibility of bulk Ce-containing catalysts can be controlled by the developed method and governs their activity in the above-mentioned reactions too. The established strategy for tuning redox properties of vanadia and ceria can be used for purposeful preparation of other metal oxides for catalytic or other material-property-related applications.
Acknowledgments
The group of Shaanxi Normal University is thankful for financial support from the National Natural Science Foundation of China (grant number 21636006). Guo-Qing Yang acknowledges support from the China Scholarship Council. Financial support by the State of Mecklenburg-Vorpommern for Leibniz-Institut für Katalyse e.V is gratefully acknowledged. The authors gratefully acknowledge the efforts of Frau Laura Kraußer for translating the English original manuscript into German. Open Access funding enabled and organized by Projekt DEAL.
Conflict of interest
The authors declare the following competing financial interest. A patent (202110110652.0) partially based on the results of this work have been issued to Shaanxi Normal University.
Open Research
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.

