

Cobalt-Catalyzed Wagner–Meerwein Rearrangements with Concomitant Nucleophilic Hydrofluorination
Graphical Abstract
A cobalt-catalyzed reaction has been developed, which proceeds via hydrogen atom transfer and radical-polar crossover steps, to form a phenonium ion under non-acidic conditions. This cationic intermediate can be regioselectively fluorinated by a tetrafluoroborate anion to give 1,2-aryl migrated products. Evaluation of distinctly substituted phenonium ion intermediates demonstrated the constraints for good reactivity and high selectivity.

Abstract
We report a cobalt-catalyzed Wagner–Meerwein rearrangement of gem-disubstituted allylarenes that generates fluoroalkane products with isolated yields up to 84 %. Modification of the counteranion of the N-fluoropyridinium oxidant suggests the substrates undergo nucleophilic fluorination during the reaction. Subjecting the substrates to other known metal-mediated hydrofluorination procedures did not lead to observable 1,2-aryl migration. Thus, indicating the unique ability of these cobalt-catalyzed conditions to generate a sufficiently reactive electrophilic intermediate capable of promoting this Wagner–Meerwein rearrangement.
Introduction
Carbocations are synthetically useful reactive intermediates as they can enable structural modifications of a molecule's carbon framework.1 For example, the generation of a carbocation that triggers the [1,2]-shift of an adjacent C−H or C−C bond to create a more stable carbocation is known as the Wagner–Meerwein rearrangement (Figure 1A).2 The formation of these positively-charged reactive intermediates can be implemented from a variety of starting materials, but often involves harsh reagents.3 Moreover, the synthetic utility of the Wagner–Meerwein rearrangement is often hampered by the presence of competing migration pathways.2 Equally, rendering these processes stereoselective is also challenging due to the low energetic barriers associated with carbocation rearrangement steps,4 although reactions that generate oxocarbenium or iminium ions have successfully led to the development of catalytic enantioselective pinacol,5 semipinacol,6 α-ketol,7 and α-iminol rearrangements.8 Exerting control over bond migrations would be synthetically powerful as the resulting changes to the carbon skeleton can provide rapid and selective access to more complex molecules.9

(a) The rearrangement of carbocations. (b) Intermediates that emulate carbocation behavior. (c) Co(IV) induced 1,2-aryl migration.
To overcome these challenges the application of carbocation surrogates that mimic the electrophilic reactivity of these intermediates has proven fruitful (Figure 1B). For example, aryliodonium(III) ions have been described as hypernucleofuges due to their extraordinary leaving group aptitude.10 These species can be readily introduced into olefin-bearing substrates using hypervalent iodine reagents. Their ability to mimic carbocation reactivity has been demonstrated in several different reactions involving structural rearrangements and anchimeric assistance.11 The prominent electrophilicity of these intermediates has been illustrated by their ability to generate phenonium ions even with electron-poor aryl rings.12 Subsequent advances have shown that phenonium ions can also be created under catalytic conditions whereby iodoarenes are oxidised in situ.13-18 This approach presents further benefits as these catalytic processes can be rendered enantioselective as the aryliodonium(III) entity is covalently bound to the substrate. Thus, it can function as a stereodefined carbocation surrogate as shown in a catalytic enantioselective Wagner–Meerwein rearrangement of β-substituted styrenes.19
More recently, alkylcobalt(IV) intermediates have also been shown to demonstrate similarly high levels of electrophilicity. These organocobalt species are formed via hydrogen atom transfer from a metal hydride to an alkene and an oxidative radical–polar crossover event. Initial studies have focused on the intra- and intermolecular displacement of the cobalt complex with various nucleophilic functional groups.20-31 The ability of alkylcobalt(IV) intermediates to acts as carbocation surrogates has been demonstrated by catalyst-controlled product-selective reactions of tertiary allylic alcohol substrates.32 The stereodefined character of the carbon bound to the cobalt(IV) complex and the use of chiral non-racemic salen ligands has enabled catalytic enantioselective transformations involving alkylcobalt(IV) species.29, 31, 33, 34 Although the electrophilic nature of alkylcobalt(IV) complexes has mostly been demonstrated in the context of nucleophilic displacements its utility as a carbocation surrogate to enable [1,2]-shifts has only been demonstrated in select semipinacol rearrangements.32, 33 Herein, we show that carbon–cobalt(IV) bonds are sufficiently electrophilic to promote 1,2-aryl migration, likely via a phenonium ion intermediate,35 without heteroatom stabilization of the incipient carbocation. Subsequent regioselective ring opening with a nucleophile restores the aromaticity and affords the rearranged product. The reaction conditions are relatively mild and non-acidic offering improved chemoselectivity and functional group tolerance when compared to alternative methods for generating free carbocations from olefins.
Results and Discussion
Determining the Optimal Reaction Conditions
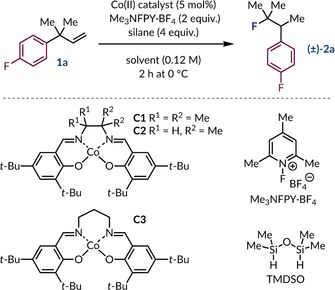
Alkene 1 a was chosen as the model substrate for the optimization phase as it enables straightforward quantification of the reaction components by 19F NMR spectroscopy (see Supporting Information for details). The preliminary conditions (Table 1, entry 1) of CoII catalyst C1 (5 mol%), N-fluoro-2,4,6-trimethylpyridinium tetrafluoroborate (Me3NFPY ⋅ BF4, 2 equiv.), 1,1,3,3-tetramethyldisiloxane (TMDSO, 4 equiv.) in PhCF3 (0.12 M) afforded the desired rearranged fluoroalkane (±)-2 a in a 38 % yield. Evaluation of various cobalt catalysts uncovered the importance of reducing the level of substitution on the ethylenediamine backbone of the salen ligand, with catalyst C2 proving optimal (Table 1, entry 2). Notably, the reaction performed very poorly if the chain length between the nitrogen atoms of the cobalt complex was increased by a methylene unit (Table 1, entry 3). Variation of the silane showed minor impact on product formation with PhMe2SiH performing narrowly better than TMDSO (Table 1, entries 4–6). A survey of reaction media showed a strong preference for aromatic solvents such as PhCF3 and PhCl, whereas other common solvents such as MeCN, CH2Cl2, and THF performed poorly (Table 1, entries 7–10). Conducting the reaction at temperatures above and below 0 °C showed a significant reduction in yield of the desired fluoroalkane (±)-2 a (Table 1, entries 11–12). Lowering the catalyst loading of C2 from 5 mol% to 3 mol% gave a minor boost in product yield (Table 1, entries 13–14). In the end, TMDSO was used in place of PhMe2SiH, with only a slight cost in yield, as byproducts derived from PhMe2SiH contaminated fluoroalkane (±)-2 a after purification by column chromatography.
|
|||||
Entry[a] |
CoII Catalyst |
Silane |
Solvent |
1 a [%][b] |
2 a [%][b] |
|---|---|---|---|---|---|
1 |
C1 |
TMDSO |
PhCF3 |
33 |
38 |
2 |
C2 |
TMDSO |
PhCF3 |
5 |
80 |
3 |
C3 |
TMDSO |
PhCF3 |
48 |
3 |
4 |
C2 |
PhSiH3 |
PhCF3 |
6 |
68 |
5 |
C2 |
PhMeSiH2 |
PhCF3 |
4 |
73 |
6 |
C2 |
PhMe2SiH |
PhCF3 |
6 |
83 |
7 |
C2 |
PhMe2SiH |
MeCN |
16 |
4 |
8 |
C2 |
PhMe2SiH |
CH2Cl2 |
43 |
38 |
9 |
C2 |
PhMe2SiH |
THF |
44 |
28 |
10 |
C2 |
PhMe2SiH |
PhCl |
3 |
87 |
11[c] |
C2 |
PhMe2SiH |
PhCl |
38 |
45 |
12[d] |
C2 |
PhMe2SiH |
PhCl |
20 |
47 |
13[e] |
C2 |
PhMe2SiH |
PhCl |
3 |
92 |
14[e] |
C2 |
TMDSO |
PhCl |
<1 |
90(76)[f] |
- [a] Unless otherwise stated optimization reactions were performed with 1 a (0.20 mmol), Me3NFPY ⋅ BF4 (2.0 equiv.), silane (4.0 equiv.) for 2 h at 0 °C. [b] Yield determined by 19F NMR analysis of the crude reaction with PhF (1.0 equiv.). [c] Reaction performed at -10 °C. [d] Reaction performed at RT. [e] 3 mol% of CoII catalyst. [f] Isolated yield in brackets. See Supporting Information for further details on optimization.
Scope and Limitations
With the optimal conditions in hand, we then explored the scope of gem-dimethyl substrates by varying the sterics, electronics, and position of the aryl ring substituents, 1 a–1 v (Scheme 1). In general, para-substitution with electronically diverse functional groups was well tolerated ((±)-2 a–2 i). The simpler systems containing no substitution, or an alkyl fragment generated the desired rearranged fluoroalkanes (±)-2 b and (±)-2 c in great yield (70–84 %). The introduction of an electron-rich methoxy group was also well tolerated giving product (±)-2 d in 68 % isolated yield. A biphenyl analogue gave 74 % yield of the rearranged fluoroalkane ((±)-2 e) and when run on a 3.0 mmol scale, from a 0.20 mmol scale, the reaction saw no discernible drop in isolated yield. Importantly, single crystal X-ray diffraction of (±)-2 e confirmed that 1,2-aryl migration had occurred.36 Halogen-containing substrates performed efficiently and chemoselectively in the presence of catalyst C2 to deliver the desired rearranged products (±)-2 a, (±)-2 f, and (±)-2 g. Remarkably, given the challenges associated with phenonium ion formation with electron-poor arenes the successful rearrangement of systems containing trifluoromethyl ((±)-2 h) and nitro ((±)-2 i) groups was surprising.37 This indicates that the (salen)cobalt(IV) complex can function as a hypernucleofuge akin to aryliodonium(III) ions.12-18 When substituents were located at the meta-position of the arene the reaction still performed superbly with halogens ((±)-2 j and (±)-2 k), electron-rich ((±)-2 l) and electron-deficient moieties ((±)-2 m). In contrast, ortho-functionalized aryl groups led the reaction to become sluggish thus resulting in longer reaction times ((±)-2 n) and lower yields of rearranged products ((±)-2 o). Additionally, the presence of an ortho-chloro substituent led to a significant amount of a benzylic fluoride ((±)-2 pb) byproduct, which was isolated independently from the desired fluoroalkane ((±)-2 pa). Multi-substituted arenes bearing two or more fluorine atoms or methoxy groups also furnished the [1,2]-aryl migrated products in 45–72 % isolated yields ((±)-2 q–2 t). Exchanging the phenyl ring for a heterocyclic thiophene afforded product (±)-2 u in 77 % yield. Unfortunately, the introduction of a Lewis basic pyridine ring ((±)-2 v) appeared to shutdown the reaction with only starting material returned.

Substrate scope for Wagner–Meerwein reaction with (a) gem-dimethyl and (b) cyclic substrates. [a] Reactions performed on a 0.20 mmol of alkene, C2 (3 mol%), Me3NFPY ⋅ BF4 (2.0 equiv.), and TMDSO (4.0 equiv.) in PhCl (0.12 M) for 2 h at 0 °C. Yields refer to isolated products while 19F NMR yields are given in parentheses (determined by 19F NMR analysis of the crude reaction mixture using PhF (1.0 equiv.) as an internal standard). [b] Reaction performed for 4 h. [c] Reaction performed with 3.0 mmol of alkene. [d] Reaction performed with 0.40 mmol of alkene. [e] Reaction performed with 0.50 mmol of alkene, 5 mol% of C2, and PhCl (0.10 M). [f] Reaction performed with 0.70 mmol of alkene. [g] Reaction performed with Me3NFPY ⋅ OTf
Next, our attention turned towards cyclic substrates where the gem-dialkyl group was constrained in various ring systems (3 a–3 e). When subjected to the reaction conditions cyclopropyl substrate 3 a did not afford the rearranged fluoroalkane (±)-4 aa, but instead generated a notable quantity of alkane 4 ab through olefin hydrogenation. This was unexpected as this was not a significant byproduct with the gem-dimethyl substrates and alkene hydrogenation has shown to be a minor pathway in closely related Co(salen) catalyzed systems.38 Likewise, cyclobutane analogue 3 b did not undergo a 1,2-aryl shift to form fluoroalkane (±)-4 ba, but generated ring-expanded cyclopentene 4 bb as the major product.39, 40 Increasing the ring size further restored selective migration of the arene albeit in poor yield for the cyclopentane product (±)-4 ca. The incorporation of six-membered rings better facilitated the rearrangement process but the yields of the fluorinated products (±)-4 da and (±)-4 ea were moderate to poor. This can be attributed to the formation of alkenes (±)-4 db and (±)-4 eb, resulting from E1 elimination post-migration, which hampers product isolation. Attempts to hydrofluorinate these olefin products (4 db and 4 eb) by introducing additional reagents post-reaction or resubjecting the pure material to the standard conditions were unsuccessful. However, performing the reaction with Me3NFPY ⋅ OTf instead of Me3NFPY ⋅ BF4 gave alkene (±)-4 db as the sole product in 71 % NMR yield.
Subsequently, we investigated a range of substrates with distinct substitution patterns to aid determination of the requirements for reactivity and selectivity (Scheme 2). A simple allylbenzene system (5 a) did not favour 1,2-aryl migration, but surprisingly the major product observed was benzyl fluoride (±)-6 aa with only trace amounts of fluoroalkane (±)-6 ab produced (Scheme 2A).41 We believe that hydrofluorination of the styrene produced via an initial Co-mediated isomerization of 5 a gives rise to product (±)-6 aa.38 Introducing a single ethyl substituent at the benzylic position restored the migratory behaviour as (±)-5 b generated rearranged fluoroalkane (±)-6 ba, which was isolated as an inseparable 1 : 2 mixture with the regioisomeric product (±)-6 bb in a combined yield of 74 %. This likely reflects the lack of an electronic bias in the phenonium ion intermediate with sterics dictating the major regioisomer (Scheme 2B).42, 43 We also investigated starting materials containing more substituted C=C bonds. For example, exposing alkene (Z)-5 c to the standard reaction conditions generated no fluoroalkane products with the alkene undergoing partial isomerization (E : Z=97 : 3 → 83 : 17), demonstrating that 1,2-dialkyl-substituted alkenes are not viable substrates under these conditions (Scheme 2C). Limited reactivity and regioselectivity was observed with 1,1-disubstituted alkene 5 d as it generated a 2 : 1 mixture of regioisomers (6 da : 6 db) in a combined NMR yield of 36 % (Scheme 2D). In contrast, 3,3,3-triphenylpropene 5 e demonstrated excellent reactivity with the major product (6 ea) resulting from elimination after phenyl migration (Scheme 2E). Low quantities of the rearranged fluoroalkane (±)-6 eb were detected by 19F NMR spectroscopy, but its poor stability reflects the preferential formation of 6 ea and precluded its isolation. We also probed the ionic nature of the reaction by using phenolic alkene 5 f, which generated dihydrobenzofuran products (±)-6 fa and (±)-6 fb in a 2.6 : 1.0 ratio via intramolecular etherification, which for the latter occurs after 1,2-aryl migration (Scheme 2F).44 This demonstrates that carbocation-like character is expressed at both the carbon atoms that form the new C−O bond during the course of the reaction.

Evaluating substrates with (a–b) a secondary or tertiary benzylic center, (c–d) 1,1- and 1,2-disubstituted alkenes, (e) a trityl group, and (f) ortho-phenol group. [a] Performed under standard conditions: 0.40 mmol of alkene, C2 (3 mol%), Me3NFPY ⋅ BF4 (2.0 equiv.), and TMDSO (4.0 equiv.) in PhCl (0.12 M) for 2 h at 0 °C. Yields refer to isolated products while 19F NMR yields are given in parentheses (determined by 19F NMR analysis of the crude reaction mixture using PhF (1.0 equiv.) as an internal standard). [b] 0.60 mmol of alkene. [c] 0.20 mmol of alkene.
To gain further insight into the nature of the migration pathway we used alternative metal-mediated hydrofluorination methods that have been proposed to only involve radical intermediates. For example, the treatment of alkene 1 a with stoichiometric iron(III) oxalate and SelectfluorTM led only to hydrofluorination of the olefin without 1,2-aryl migration and provided fluoroalkane (±)-7 in 34 % yield (Scheme 3A).45 Similarly, a nickel-catalyzed protocol that employs N-fluorobenzenesulfonimide (NFSI) did not generate the rearranged fluoroalkane (±)-2 a, nor any another rearranged compounds previously observed under the cobalt-catalyzed conditions, but solely product (±)-7.46 Given the evidence for carbocation-like reactivity in cobalt-catalyzed processes that use N-fluoropyridinium salts,20, 32, 47 and the lack of aryl migrated products with the other hydrofluorination protocols tested suggests that the reaction proceeds via a phenonium ion intermediate.42 Thus, a neophyl-type radical rearrangement pathway for aryl migration seems less likely.48 Additionally, to test for ionic reactivity in the absence of any metals we treated neat 1 a with pyridine ⋅9HF (10 equiv.), but no reactivity was observed with near full recovery of 1 a after 5 hours (96 %).49

(a) Evaluation of different hydrofluorination methods. (b) The role of the oxidant counterion. Yields determined by 19F NMR analysis of the crude reaction mixture using PhF (1.0 equiv.) [a] Reaction conditions as per Table 1, entry 14. [b] Iron-mediated radical hydrofluorination.45 [c] Nickel-catalyzed radical hydrofluorination.46 [d] pyridine ⋅9HF (10 equiv.), neat, RT, 5 h. [e] Performed with 0.10 mmol of alkene 1 a, C2 (3 mol%), Me3NFPY ⋅ OTf (2.0 equiv.), additive (2.0 equiv.) and TMDSO (4.0 equiv.) in PhCl (0.12 M) for 2 h at 0 °C.
To determine the source of the fluorine in product (±)-2 a we employed an N-fluoropyridinium oxidant with a triflate anion rather than tetrafluoroborate counterion. This led to a noticeable change in product composition as fluoroalkane (±)-2 a was no longer observed, but instead alkenes 8 and (±)-9 were obtained by elimination from the phenonium ion (Scheme 3B, Entry 1). Reintroducing tetrafluoroborate anions into the reaction partially restored the formation of the fluorinated product (±)-2 a. The use of NaBF4 as an additive with Me3NFPY ⋅ OTf had minimal impact on the product distribution likely due to its low solubility in chlorobenzene (Scheme 3B, Entry 2). Switching to a more lipophilic tetrabutylammonium cation resolved this issue and fluoroalkane (±)-2 a was again the major product implying that the tetrafluoroborate anion is acting as a nucleophilic fluoride source (Scheme 3B, Entry 3).50
Based on prior work and our own findings we tentatively present the following catalytic cycle for the generation of the phenonium ion intermediate (Figure 2). The combination of cobalt(II)-salen catalyst A, silane, and Me3NFPY ⋅ BF4 is proposed to generate a transient CoIII−H species B and a cationic CoIII complex C.51 Hydrogen atom transfer from the metal hydride (MHAT) to the olefin 1 b would generate a carbon-centred radical solvent-caged with the CoII catalyst A.52 At this stage cage collapse would form organocobalt(III) complex D, which appears to resist rearrangement. Subsequent, single electron oxidation (SEO) of the alkylCo(III) species D could be effected by the cationic CoIII complex C or by the excess of oxidant.23, 47 The produced alkylcobalt(IV) intermediate E, which acts as a carbocation surrogate, could incite formation of a phenonium ion and lead to regeneration of the CoII catalyst A. Preferential 1,2-migration of the aryl group over the alkyl group is consistent with their migratory aptitude in carbocation instigated rearrangements.53 The tetrafluoroborate anion could then regioselectively fluorinate the phenonium ion to give the rearranged product (±)-2 b.54

Proposed catalytic cycle for the reaction.
Conclusion
In summary, we have developed a cobalt-catalyzed Wagner–Meerwein hydrofluorination protocol that utilizes a simple CoII-salen catalyst and Me3NFPY ⋅ BF4 as a bifunctional reagent that acts as an oxidizing agent and fluoride source. This mild, general, and scalable procedure allows for synthesis of fluoroalkanes in good to excellent yield and with high functional group tolerance. Our experimental evidence supports a cationic reaction pathway whereby a phenonium ion intermediate is generated, which is regioselectively fluorinated, to give products resulting from 1,2-migration of the aryl ring.
Acknowledgments
The authors acknowledge funding from the Royal Society (University Research Fellowship URF\R1\180017 (CPJ) and associated Enhancement Award RGF\EA\181022 (CPJ and RHH)), and the EaSI-CAT Centre for Doctoral Training (RHH and NM). We thank Prof. Andy Smith, Prof. Allan Watson, and their respective research groups for sharing their lab equipment and chemical inventories.
Conflict of interest
The authors declare no conflict of interest.
Open Research
Data Availability Statement
The research data underpinning this publication can be accessed at https://doi.org/10.17630/5944443f-21ce-4570-95c4-ee55c518f9c1.

