

Hochgradig enantioselektive Organokatalyse mit zweizähnigen Halogenbrückendonoren
Zusammenfassung
Da sich der Einsatz „nicht-klassischer“ nicht-kovalenter Wechselwirkungen wie der Halogenbrücken (XB) in der asymmetrischen Katalyse noch in einem sehr frühen Stadium befindet, gibt es noch erhebliche Herausforderungen zu überwinden. In einigen berichteten Beispielen ist die Bedeutung der Halogenbrücken für die katalytische Wirkung unklar, während in anderen Fällen die Katalysatoraktivität begrenzt ist. Wir stellen hier die zweite Generation eines zweizähnigen Iod(I)-basierten Halogenbrückendonors als modifizierbaren und hochaktiven chiralen Halogenbrückenkatalysator vor. Mit diesen modifizierten Derivaten konnte in einer Mukaiyama-Aldol-Modellreaktion für eine Reihe verschiedener Substrate eine hohe Stereokontrolle von bis zu 98 % ee erreicht werden. Wichtig ist hierbei, dass die entscheidende Rolle der Halogenbrücken in dieser Katalyse durch die geringe Leistung der nicht-iodierten Varianten und durch DFT-Berechnungen untermauert wurde. Letztere deuten auch darauf hin, dass die Stereoinduktion auf der auferzwungenen Orientierung der Substrate zueinander beruht.
Im Laufe der letzten paar Jahrzehnte hat sich die nicht-kovalente Wechselwirkung der Halogenbrücken (XB)[1-7] von einem eher obskuren zu einem fast routinemäßigen Konzept für viele Anwendungen entwickelt, z. B. im Crystal Engineering,[8-14] in der biomolekularen und medizinischen Chemie,[15-19] sowie in der supramolekularen Erkennung von Lewis-Basen (LB) in Lösung.[20-24] Weitere Forschungsarbeiten haben auch das Potential von Halogenbrückendonoren zur Beschleunigung organischer Reaktionen aufgezeigt, wodurch sie faszinierende Alternativen zu etablierten Organokatalysatoren bieten.[25-28]
Obwohl inzwischen eine Vielzahl an Reaktionen[29-47] von Halogenbrückendonoren katalysiert oder aktiviert wurden, ist es nach wie vor schwierig, mit dieser Wechselwirkung eine asymmetrische Induktion zu erreichen.[48] Die intrinsischen Merkmale der Halogenbrücken, nämlich die starke Direktionalität der R-X–LB Wechselwirkung sowie der große Abstand des Substrats zum (chiralen) Katalysatorgrundgerüst, stellen erhebliche Herausforderungen für die Entwicklung einer wirksamen Enantiokontrolle dar. Folglich wurde erst in den letzten fünf Jahren über erste Erfolge in diesem Bereich berichtet.
Im Jahr 2020 stellte unsere Gruppe einen chiralen bis(imidazolium)-basierten Katalysator (1BArF) vor, der in einer Mukaiyama-Aldolreaktion nur mäßige Enantioselektivität erreichen konnte (Abbildung 1).[49] Später im selben Jahr gaben Taylor, Garcia-Mancheño und Mitarbeiter die Anwendung eines neutralen vierzähnigen iodtriazol-basierten Systems zur Anionenbindungskatalyse bekannt, mit ähnlich begrenzter Selektivität.[50] 2023 konnten sie jedoch Enantiomerenüberschüsse von bis zu 90 % ee erreichen, allerdings nur mit bestimmten Substraten, welche zusätzliche elektrophile Substituenten aufwiesen.[51]

Zudem veröffentlichten Yoshida und Mitarbeiter mehrere Beispiele, in denen bifunktionale Halonium(III)-Salze als wirksame und enantioselektive Katalysatoren in der Reaktion von Isatinderivaten mit verschiedenen Nukleophilen agieren. Allerdings scheinen Halogenbrücken in diesen Fällen eher eine ergänzende Rolle zu spielen.[52, 53] Hohe Enantiomerenüberschüsse in dieser Reaktion wurden kürzlich auch von Nachtsheim präsentiert, hierbei wurden einzähnige Iod(III)-Derivate eingesetzt.[54]
Somit gibt es derzeit noch keinen Präzedenzfall für die hoch enantioselektive Aktivierung von nicht angepassten Substraten durch Iod(I)-basierte Katalysatoren oder durch zweizähnige Katalysatoren im Allgemeinen, bei denen Halogenbrücken als entscheidende Ursache („Motor“) fungieren. Wir stellen hier ein erstes solches Beispiel vor, bei dem eine stärkere, modifizierbare Variante unseres früheren Katalysatormotivs 1BArF Enantioselektivitäten von mehr als 90 % induziert.
Hierzu sollte nach unseren Überlegungen bei der Synthese eines modifizierbaren Katalysatorsystems die Derivatisierung eines gemeinsamen Vorläufers so spät wie möglich erfolgen. Wegen der Verträglichkeit gegenüber verschiedenen Reaktionsbedingungen und der Fülle an Möglichkeiten für eine spätere Derivatisierung wurde daher das leicht zugängliche[55] (1R,2S)-1-Amino-6-brom-indan-2-ol (2) als Ausgangspunkt gewählt (Schema 1).

Von hier aus erfolgte die Umwandlung zum N-Formylmethylketon 3 in 5 Schritten in 24 % Ausbeute, basierend auf etablierten Verfahren (siehe SI).[56] Dieses Keton wurde dann zur Bildung der entsprechenden Bis(imidazolium)-Spezies 5aCl (Schema 1) in 53 % Ausbeute in einem Eintopfverfahren über drei Schritte verwendet, analog zu den für die Synthese von 1BArF entwickelten Methoden.
Da sich die Derivatisierung dieses Imidazolium-Zwischenprodukts in orientierenden Studien als schwierig erwies, wurde eine weitere Modifikation bereits auf der Stufe des robusteren Ketons 3 vorgenommen. Hierbei wurde durch eine Ullmann-artige Kupplung[57] ein Trifluoracetamidrest als Ankerpunkt eingeführt, welches später unter milden Bedingungen modifizierbar sein sollte. Das hieraus resultierende Zwischenprodukt 4 konnte in drei Schritten mit 49 % Ausbeute in das Bis(imidazolium)-Derivat 5bCl umgewandelt werden. Nach Abspaltung des Trifluoracetamids wurde das breit funktionalisierbare Anilin 6Cl in 85 %iger Ausbeute erhalten, welches spätere Modifikationen ermöglichen sollte. Als erste Derivate wurden Amide wie 5c–eCl ins Auge gefasst: sie ermöglichen eine stabile Verknüpfung der neuen Substituenten und erlauben einen direkten Vergleich mit der ursprünglichen Struktur 5bCl. Alle diese Verbindungen wurden dann mit N-Iodsuccinimid (NIS) glatt in Halogenbrückendonoren umgewandelt. Der schrittweise Anionenaustausch und die (teilweise) Trennung der Atropisomere führte zu den starken, funktionalisierten Halogenbrückendonoren 7b–eCl. Die Trennung der Atropisomere erwies sich zunächst als schwierig, und in der Regel wurden Gemische erhalten. Alle im Folgenden berichteten Screening-Experimente wurden daher mit diesen Mischungen durchgeführt, bei denen sehr wahrscheinlich nur das syn-Atropisomer die aktive Spezies darstellt. Ein Vergleich der Enantiomerenüberschüsse der verschiedenen Katalysatoren sollte dennoch valide sein, was später für 7bBArF bestätigt wurde, nachdem das reine „syn“-Isomer erhalten werden konnte (siehe unten).
Die erzielten Ausbeuten können jedoch nur als Schätzung der Katalysatorleistung angesehen werden, da das aktive syn-Atropisomer in unterschiedlichem Maße mit dem entsprechenden anti-Isomer „verdünnt“ ist. Ähnliche Verfahren zur Iodierung und zum Anionenaustausch wurden auch für die Bromo-Spezies 5aCl angewandt, wodurch die XB-Katalysatorstruktur 7aBArF erhalten wurde. Um die Bedeutung der Halogenbrücken im Verhältnis zu einer möglichen zusätzlichen Wasserstoffbrücke (vom Katalysatorrückgrat aus) zu bestimmen, wurden auch die H-Analoga 5aBArF und 5bBArF hergestellt (siehe SI).
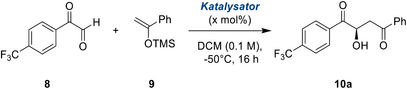
Mit dieser Ausgangscharge an modifizierten, zweizähnigen Halogenbrückendonoren testeten wir ihre Leistungsfähigkeit in der asymmetrischen Mukaiyama-Aldolreaktion von Arylglyoxalen, in der wir zuvor nur eine mäßige Enantioselektivität erzielen konnten. Als Modellsubstrat wählten wir die wenig erforschte[58] trifluormethyl-tragende Variante 8 (Tabelle 1, oben), welche eine einfache Verfolgung der Reaktion mittels 19F-NMR erlaubt. Während orientierender Studien erwies sich die Verwendung von wasserfreien Arylglyoxalen als entscheidend für eine hohe Selektivität und einen hohen Umsatz; allerdings neigen die monomeren Arylglyoxale zur Oligomerisierung, was zu geringeren Ausbeuten führt. Nach einigen Optimierungen für dieses Substrat konnte das Produkt jedoch mit gut reproduzierbaren Enantioselektivitäten und akzeptablen Ausbeuten gewonnen werden.
 |
||||
| Nr. | Katalysator | Katalysatorladung [mol%] | Ausbeute [%]b) | ee [%]c) |
|---|---|---|---|---|
| 1 | 1BArF | 5 | 51 | 67 |
| 2 | 7aBArF | 5 | 51 | 77 |
| 3 | 7bBArF | 5 | 45 | 94 |
| 4 | 7cBArF | 5 | 46 | 91 |
| 5 | 7dBArF | 5 | 29 | 81 |
| 6 | 7eBArF | 5 | 9 | 11 |
| 7 | 5aBArF | 5 | 6 | 13 |
| 8 | 5bBArF | 5 | 12 | <2 |
| 9 | 7bBArF | 2.5 | 25 | 94 |
| 10 | 7bBArF | 10 | 50 | 94 |
| 11d) | “syn”-7bBArF | 5 | 63 | 94 |
| 12d) | “syn”-7bBArF | 1.7 | 50 | 94 |
| 13d) | “syn”-7bBArF | 0.8 | 43 | 93 |
- a) Alle Reaktionen wurden im Maßstab von 0.09 mmol in trockenem CH2Cl2 unter Argon-Atmosphäre mit 2 Äquivalenten Silylenolether 9 durchgeführt.
- b) Isolierte Ausbeuten.
- c) Bestimmt durch chirale HPLC-Analyse.
- d) Mit einem reinen Atropisomer des Katalysators.
Mit 5 % Katalysatorladung lieferte der unsubstituierte Katalysator 1BArF nach 16 Stunden bei -50 °C das Produkt 10a mit einer akzeptablen Enantioselektivität von 67 % ee, während der bromsubstituierte Katalysator 7aBArF bereits eine höhere Selektivität von 77 % ee erbrachte (Tabelle 1, Einträge 1 und 2). Ein weiterer deutlicher Sprung in der Stereokontrolle wurde mit der Trifluoracetamidgruppe von 7bBArF beobachtet, wodurch hervorragende 94 % ee erreicht werden konnten (Tabelle 1, Eintrag 3). Unseres Wissens nach ist dies die erste Organokatalyse, mit der bei solchen Aldolreaktionen mehr als 90 % Enantiomerenüberschuss erzielt werden konnten.[59, 60] Der vergleichsweise geringe Unterschied im Enantiomerenüberschuss zwischen den brom- und amidsubstituierten Donoren 7aBArF und 7bBArF deutet darauf hin, dass die entscheidende Enantioinduktion vom Katalysatorrückgrat ausgeht, während das möglicherweise wasserstoffbrücken-bindende Amid höchstens eine unterstützende Rolle spielt. Glücklicherweise kristallisierte das Aldolprodukt aus diesen Experimenten in enantiomerenreiner Form aus, und seine Konfiguration konnte als (R)-10a bestimmt werden (siehe SI).[61]
Der strukturell ähnliche, aber sterisch unwesentlich weniger anspruchsvolle Katalysator 7cBArF zeigte eine ähnliche Aktivität, mit einer Ausbeute von 46 % nach 16 Stunden und einer geringfügig verringerten Selektivität von 91 % ee (Tabelle 1, Eintrag 4). Derivat 7dBArF, welches im Vergleich zu 7bBArF größere sterische Hinderung aufweist, lieferte ebenfalls eine geringere Enantioselektivität (81 % ee, Tabelle 1, Eintrag 5), was auf eine gute sterische Passung des Stammderivats 7bBArF hindeutet. Interessanterweise schnitt XB-Donor 7eBArF mit seinen elektronenreicheren Isobutyramid-Gruppen schlecht ab, da nach 16 Stunden nur 9 % des Aldolprodukts erhalten wurden (Tabelle 1, Eintrag 6). Ein Vergleich mit den Ergebnissen der nicht amidsubstituierten Katalysatoren 1BArF und 7aBArF zeigt, dass die elektronenreichen Isobutyramid-Gruppen der katalytischen Aktivität abträglich zu sein scheinen, was zu drastisch niedrigerer Ausbeute und Selektivität für 7eBArF führt.
Die Rolle der Halogenbrücken sowohl für die Aktivität als auch für die Selektivität des Katalysators wurde dann durch die Verwendung der nicht-iodierten Referenzverbindungen 5aBArF und 5bBArF näher untersucht. Der bromsubstituierte Wasserstoffbrückendonor 5aBArF lieferte 6 % des Produkts mit einem Enantiomerenüberschuss von nur 13 %, und das Trifluoracetamid-Derivat 5bBArF lieferte 12 % des racemischen Produkts (Tabelle 1, Einträge 7 und 8).
All diese Ergebnisse deuten eindeutig darauf hin, dass Halogenbrücken die entscheidende Wechselwirkung dieser Katalyse darstellen, nicht nur bei der Aktivierung des Substrats, sondern auch als wesentliche Triebkraft für eine hohe asymmetrische Induktion. Die geringen Ausbeuten, die für 5aBArF und 5bBArF beobachtet wurden, sind möglicherweise das Ergebnis der Aktivierung durch Wasserstoffbrücken.
Weitere Variationen der Reaktionsbedingungen für den besten Katalysator 7bBArF ergaben keine nennenswerten Verbesserungen: während eine Katalysatorladung von 10 % zu einer nur geringfügig verbesserten Menge an Produkt führte, wurde mit 2.5 mol % eine erheblich geringere Ausbeute erzielt. In beiden Fällen wurde weiterhin die gleiche ausgezeichnete Stereoselektivität beobachtet (Tabelle 1, Einträge 9 und 10). Wie bereits erwähnt, wurden alle bisher beschriebenen Experimente mit Mischungen von syn-/anti-Atropisomeren durchgeführt. Für 7bBArF konnte schließlich das reine „syn“-Atropisomer[62] in kleinen Mengen gewonnen werden, wodurch ein Vergleich seiner Leistungsfähigkeit mit der des Gemisches möglich wurde. Während die Enantioselektivitäten unverändert blieben (Tabelle 1, Einträge 11–13), verbesserte sich die Ausbeute bei gleicher Gesamtbeladung des Katalysators merklich (Eintrag 11 gegen Eintrag 3). Die Ausbeute konnte jedoch durch Verwendung der äquivalenten Menge des reinen syn-Isomers (1.7 mol %) angenähert werden, die in der 5-mol %-Beladung des Gemisches vorhanden wäre (Eintrag 12).[63] Selbst wenn die Beladung des reinen Katalysators auf unter 1 % gesenkt wird, werden immer noch ordentliche Ausbeuten erzielt.
Daraufhin verlagerte sich unser Interesse auf ein Screening der in der Reaktion verwendeten Silylenolether. Da wir derzeit hauptsächlich an der Aufklärung des Mechanismus' der Enantioinduktion durch die Halogenbrückendonoren interessiert sind, könnte das Verhalten verschiedener Substrate Rückschlüsse auf die Struktur des entscheidenden Übergangszustandes zulassen. Überraschenderweise führten bereits leichte Veränderungen, beispielsweise die Einführung von Methylgruppen in den Produkten 10b-10d, zu einem deutlichen Verlust an Enantioselektivität (Schema 2). Während die mit 2-Me-(10b) und 3-Me-(10c) substituierten Produkte in ähnlicher Ausbeute und Selektivität erhalten wurden, war der Enantiomerenüberschuss des mit 4-Me-substituierten Aldolprodukts 10d mit nur 70 % ee sogar noch geringer. Dennoch konnten für das methoxy-substituierte Produkt 10e und das Fluoridderivat 10f immer noch 87 % und 82 % ee erzielt werden, während das Bromderivat 10g mit geringerer Selektivität erhalten wurde (Schema 2).

Interessanterweise konnte das Cyclohexyl-Analogon 10h in mäßiger Ausbeute und mit einer signifikanten Enantioselektivität von 86 % ee erhalten werden, während das Thiophen-Derivat eine geringere Selektivität aufwies (Schema 2, 10i). Andererseits wurden mit erweiterten π-Systemen beispielsweise bei den 2-Benzothiophenyl- (10j), 4-Biphenyl- (10k) und 2-Naphthyl- (10l) substituierten Aldolprodukten ausgezeichnete Enantioselektivitäten von bis zu 98 % ee beobachtet. Das Screening weiterer Nukleophile ergab, dass stark elektronenarme Nukleophile (10m und 10n) sowie Silylenolether mit erhöhtem sterischen Bedarf in der Nähe der Alkengruppen (10o bis 10r) unter den optimierten Bedingungen nicht toleriert wurden.
Während diese Ergebnisse auch einige Grenzen des Anwendungsbereichs aufzeigen, bestätigen sie gleichzeitig aber, dass mit anderen Substraten hervorragende Selektivitäten erzielt werden können und dass für einige weniger geeignete Substrate eine immerhin gute Enantioselektivität beobachtet wird. Die engen Anforderungen an die Substrate, die für eine wirklich außergewöhnliche Enantiokontrolle erforderlich sind, lassen auf eine enge Verflechtung zwischen Katalysator und Substrat im Übergangszustand schließen.
Als wir unsere Aufmerksamkeit auf die Evaluierung unterschiedlich substituierter Arylglyoxale richteten, wurde eine ähnliche Empfindlichkeit der asymmetrischen Induktion gegenüber scheinbar kleinen Änderungen des Substrats beobachtet. Obwohl das bromsubstituierte Glyoxal 10s (Schema 3) mit 94 % ee in das Aldolprodukt umgewandelt werden konnte – ein weiterer Fall von sehr hoher Enantioselektivität -, erwies sich der Versuch als schwierig, die besonderen Eigenschaften eines weiteren Substituenten zu ermitteln, welcher solch ein Maß an Stereokontrolle ermöglicht: ein Isopropylsubstituent, der als Isoster der Trifluormethylgruppe angesehen wird,[64-67] führte zu 51 % Ausbeute, aber nur 75 % ee für das Aldolprodukt 10t.

Auch der Versuch, die elektronenziehenden Eigenschaften[68] der Trifluormethylgruppe mit Hilfe eines Cyanosubstituenten grob nachzuahmen, führte nur zu einer Selektivität von 73 % ee für das Produkt 10u. Andererseits wurde das fluor-substituierte Produkt 10v mit etwas höherer Selektivität erhalten, während das nicht-substituierte Produkt 10w mit nur 67 % ee isoliert wurde.
Dennoch konnte auch für den Ester 10x und den Thioether 10y eine außergewöhnliche Enantioselektivität beobachtet werden, jeweils mit Enantioselektivitäten von 97 % ee bzw. 98 % ee. Während die möglicherweise konkurrierende nukleophile Estergruppe von 10x toleriert wurde, beeinträchtigte die Einführung eines Acetamids (in Produkt 10z) die Produktbildung erheblich und führte auch zu einer geringeren Enantioinduktion. Zusammen mit der Unverträglichkeit des methylierten Anilins in der Reaktion zu 10aa deutet dies auf Einschränkungen des beschriebenen Verfahrens in Gegenwart konkurrierender Nukleophile hin.
Aufgrund der Komplexität des Katalysators ist es nicht trivial, die Schlüsselmerkmale zu verstehen, die zu einer so hohen Enantioselektivität führen. Wir setzten daher die Computerchemie ein, um Einblicke in die Stereoinduktion bei der stereobestimmenden C–C-Bindungsbildung der Aldolreaktion zu erhalten. Für die Berechnungen wurden das Substrat 8 und der Enolether 9 sowie der dikationische Katalysator 7b verwendet. Mit Hilfe von CREST wurde ein umfassendes Sampling der Konformere der Übergangsstrukturen durchgeführt, die zu den (R)- und (S)-Enantiomeren von 10a führen.[69, 70] Um ein vollständiges Sampling zu gewährleisten, wurden beide prochiralen Seiten des Substrats 8 und beide diastereotopen Seiten des Enolethers 9 untersucht. Die verschiedenen Konformere wurden mittels M06-2X/def2-TZVP (def2-TZVPD für I)//M06-2X/def2-SVP (def2-SVPD für I) modelliert, wobei das SMD-Lösungsmittelmodell für Dichlormethan (mit überarbeiteten Radien für Iod) verwendet wurde.[71] Für jeden Übergangszustand, der zu den (R)- und (S)-Produkten führt, wurden zwei Konformere identifiziert, die zu über 99 % zur Boltzmann-Population beitragen (siehe SI). Die stabilsten Konformere der Übergangszustände (TS-1R und TS-1S) machen jeweils mehr als 80 % der Boltzmann-Populationen aus und sind in Abbildung 2a dargestellt.

Der berechnete Unterschied an freier Enthalpie zwischen den Übergangszuständen beträgt 3.5 kJ/mol zugunsten von (R). Bei −50 °C entspricht dies einer vorhergesagten Enantioselektivität von 85 % ee, was gut mit der experimentell beobachteten Selektivität von 94 % ee übereinstimmt. Außerdem liegen die Aktivierungsbarrieren des katalysierten Prozesses bei 31.4 kJ/mol (R) bzw. 34,9 kJ/mol (S) und damit deutlich unter der des unkatalysierten Prozesses (51.2 kJ/mol, siehe SI), wodurch die beschleunigende Wirkung des Katalysators unterstrichen wird. Bemerkenswerterweise ist in beiden Übergangszuständen das Substrat auf die gleiche Art und Weise gebunden: die beiden Iodzentren chelatisieren die Carbonylgruppe des Aldehyds, wobei eines der Iodatome zusätzlich eine schwächere Halogenbrücke zum zweiten Carbonyl-Sauerstoff ausbildet. In beiden Übergangszuständen fungiert der Sauerstoff einer Amidgruppe als Wasserstoffbrückenakzeptor – entweder gegenüber dem Aldehyd (TS-1R) oder dem Silylenolether (TS-1S) –, beide Wechselwirkungen sind allerdings relativ schwach ausgeprägt. Trotz mehrfacher Versuche konnte in keinem der beiden Fälle ein energetisch sinnvolles Konformer gefunden werden, in dem das Amid über das NH-Wasserstoffatom als Wasserstoffbrückendonor fungieren würde.
Ein wesentlicher Unterschied zwischen den beiden Übergangszuständen liegt in ihrer Position entlang der Reaktionskoordinate: TS-1S stellt einen späteren Übergangszustand dar, mit einem C–C-Abstand der sich bildenden Bindung von 1.88 Å im Vergleich zu 2.10 Å bei TS-1R. Dies zeigt sich auch in der Wechselwirkung mit dem Sauerstoffatom des Aldehyds, wobei Abstände von 2.81 Å und 2.60 Å beobachtet werden, im Gegensatz zu 2.91 Å und 2.69 Å bei TS-1R. Dies weist auf eine stärkere Wechselwirkung hin, bedingt durch die erhöhte Elektronendichte am Sauerstoffatom der Aldehyd-Carbonylgruppe, welche vom eintretenden Nukleophil induziert wird. Angesichts der komplexen Natur der Übergangszustände ist es jedoch schwierig, ein einzelnes Merkmal zu identifizieren, welches die Selektivität eindeutig erklären würde.
Daher wurde eine Distortion/Interaction-Analyse (Activation Strain Model) von TS-1R und TS-1S durchgeführt, um einen möglichen Erklärungsansatz zu finden (Abbildung 2b).[72] Bemerkenswerterweise fällt die Wechselwirkungsenergie zugunsten von TS-1S aus, was mit dem späten Charakter dieses Übergangszustands sowie der verstärken Halogenbrücken zwischen Substrat und Katalysator in diesem Fall in Einklang steht (Abbildung 2b, oranger Balken). Im Rahmen der Verzerrungsanalyse wurden die Übergangsstrukturen in Fragmente des Katalysators und des Aldol-Übergangszustands 8…9 unterteilt, woraus sich die Energien ΔEDist(Cat) und ΔEDist(TS) ergeben. Hieraus ging hervor, dass der Hauptvorteil von TS-1R aus der deutlich höheren Verzerrungsenergie ΔEDist(TS) von TS-1S resultiert (Abbildung 2b, grüner Balken). Aus diesem Grund wurden die Eigenschaften der Fragemente des Aldol-Übergangszustands 8…9 in TS-1R und TS-1S genauer untersucht; die entsprechenden Ergebnisse sind in Abbildung 3 dargestellt.

Für das Aldol wird in TS-1R eine anti-periplanare Orientierung zwischen 8 und 9 beobachtet, wodurch die Silylgruppe nach der C–C-Bindungsbildung ideal für die Übertragung positioniert ist. Diese Konformation minimiert sterische Wechselwirkungen. Im Gegensatz dazu zeigt TS-1S eine synklinale Orientierung von 8 und 9, was zu erheblichen sterischen Konflikten führt. Der aus TS-1R extrahierte Übergangszustand wurde erneut optimiert, um einen tatsächlich optimierten, unkatalysierten Übergangszustand zu erhalten. Dies ergab eine Aktivierungsbarriere von 53.2 kJ/mol, was dem niedrigstenergetischen Übergangszustand des unkatalysierten Prozesses (51.2 kJ/mol) sehr nahekommt, welcher im Übrigen ebenfalls eine anti-periplanare Orientierung aufweist (siehe SI).
Basierend auf diesen Analysen ergibt sich folgendes Bild: 1) Die doppelte Halogenbrücke an das Aldehyd-Carbonyl stellt den primären Aktivierungsmechanismus des Katalysators 7b dar. Konformationsanalysen zeigen, dass diese Bindung die Energie deutlich senkt und zur Ausbildung der energieärmsten Übergangsstrukturen führt. 2) Zwar blockiert die Wechselwirkung des Substrats mit dem Katalysator nicht effektiv eine Seite des Aldehyds, jedoch bewirkt das gesamte chirale Umfeld entfernte Einschränkungen der Orientierung des eintretenden Enolether-Nukleophils. 3) Die Selektivität ergibt sich daher in erster Linie aus der Orientierung von 8–9 im C–C-Bindungbildungsschritt der Aldolreaktion, wobei das Hauptenantiomer über die stark bevorzugte anti-periplanare Orientierung entsteht. Diese Orientierung minimiert sterische Wechselwirkungen.
Es ist derzeit noch zu früh, um die subtilen Effekte der Natur der Substrate 8 oder der Enolether 9 auf die Selektivitäten vollständig zu erfassen. Weitere Untersuchungen sind erforderlich, um den vollständigen stereochemischen Induktionsprozess sowie die Beziehung zwischen Substrat und Katalysator aufzuklären. Beim Katalysator deutet sich jedoch an, dass die verbesserten Selektivitäten von 7b gegenüber 1 auf die größere räumliche Ausdehnung der Trifluoracetamidgruppen zurückzuführen sind. Diese erzeugen ein größeres sterisches Volumen, welches die optimale Orientierung des Enolethers 9 im Aldol-Übergangszustand für das (S)-Enantiomer erschwert. Hingegen wird eine ideale Positionierung von Substrat 8 im Übergangszustand der Bildung des (R)-Enantiomers ermöglicht.
Zusammenfassend lässt sich feststellen, dass ein modifizierbarer, bidentater chiraler Halogenbrückendonor in einer Mukaiyama-Aldol-Testreaktion zu exzellenten Enantioselektivitäten führte. Neben dem optimalen Katalysator mit Trifluoracetamid-Substituenten wurden auch Varianten mit anderen Gruppen getestet: Difluoracetamid erzielte ebenfalls über 90 % Enantioselektivität, eine immer noch ordentliche asymmetrische Induktion wurde auch mit Trichloracetamid sowie mit Bromsubstituenten erreicht, wohingegen mit Isobutyramid nur ein sehr geringer Enantiomerenüberschuss beobachtet wurde.
Ein Substratscreening der Reaktionspartner offenbarte ein signifikantes Grundniveau an Stereoselektivität für verschiedene Silylenolether und Arylglyoxale, wobei selbst scheinbar kleine Variationen zu spürbar verringerter asymmetrischer Induktion führten. Kontrollversuche mit nicht-iodierten Analoga bestätigten erneut die entscheidende Rolle der Halogenbrücken in diesem Katalysatormotiv – sowohl für die Aktivität als auch für die Enantioselektivität. DFT-Berechnungen zeigten, dass die bevorzugte Bildung des (R)-Produkts auf eine ungünstige Orientierung der Aldolpartner in der Tasche des chiralen Katalysators im Fall des (S)-Produkts zurückzuführen ist.
Insgesamt stellt dies den ersten Fall dar, in dem hohe asymmetrische Induktion mit einem Iod(I)-basierten (und bidentaten) Katalysator überwiegend durch Halogenbrücken und mit allgemeinen (nicht halogenbrückenbindenden) Substraten erzielt wurde. Damit stellt diese Arbeit einen wichtigen Schritt in der Weiterentwicklung zunehmend ausgefeilterer Organokatalysen unter Nutzung dieser Wechselwirkung dar – auch wenn Anpassungen der Katalysatorstruktur an andere Substrattypen wahrscheinlich erforderlich sein werden.[73]
Hintergrundinformationen
Die Autoren haben in der Supporting Information weitere Literaturstellen zitiert.[74-98]
Danksagungen
Finanziert von der Deutschen Forschungsgemeinschaft (DFG) im Rahmen der deutschen Exzellenstrategie (EXC 2033–390677874 RESOLV) und über den Antrag HU 1782/6-1.
Open Access Veröffentlichung ermöglicht und organisiert durch Projekt DEAL.
Interessenkonflikt
Die Autoren erklären, dass keine Interessenkonflikte vorliegen.
Erklärung zur Datenverfügbarkeit
The data that support the findings of this study are available from the corresponding author upon reasonable request.

