

Sequential C−F Bond Transformation of the Difluoromethylene Unit in Perfluoroalkyl Groups: A Combination of Fine-Tuned Phenothiazine Photoredox Catalyst and Lewis Acid
Abstract
A sequential process via photoredox catalysis and Lewis acid mediation for C−F bond transformation of the CF2 unit in perfluoroalkyl groups has been achieved to transform perfluoroalkylarenes into complex fluoroalkylated compounds. A phenothiazine-based photocatalyst promotes the defluoroaminoxylation of perfluoroalkylarenes with (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) under visible light irradiation, affording the corresponding aminoxylated products. These products undergo a further defluorinative transformation with various organosilicon reagents mediated by AlCl3 to provide highly functionalized perfluoroalkyl alcohols. Our novel phenothiazine catalyst works efficiently in the defluoroaminoxylation. Transient absorption spectroscopy revealed that the catalyst regeneration step is crucial for the photocatalytic aminoxylation.
The carbon–fluorine (C−F) bond transformation in perfluoroalkyl compounds not only is an important synthetic method in organic chemistry,1 but also is an urgent issue to solve PFAS (polyfluoroalkyl substances) environmental problems.2 Numerous C−F bond activation protocols have been reported for single C−F bond transformations of perfluoroalkyl group.3, 4 However, a sequential C−F bond transformation of a difluoromethylene unit (−CF2−) into two different functional groups remains underdeveloped (Figure 1A) despite being an important clue to the solution of PFAS problems. This is because the harsh reaction conditions needed to cleave robust C−F bonds cause the undesired installation of the same functional group. In fact, dialkoxylation,5 dimethylation6 and dichlorination6 of a CF2 moiety have been reported. To avoid installing the same groups, amino alcohols were used in the aminoalkoxylation of α-perfluoroalkyl ketones in a three-component tandem reaction (Figure 1B, a).7 Recently, two distinguished reactions were reported: a sequential defluorinative alkylation of trifluoroacetyl compounds by a radical mechanism (Figure 1B, b)8 and a coupling reaction of 1,1-difluoroalkyl compounds (RCF2R’) with Grignard reagents and chlorosilanes or alkyl tosylates by CrCl2 catalysis via chromium carbenoid species (Figure 1B, c).9 Neither method is applicable to the transformation of longer perfluoroalkyl compounds (RCF2(CF2)nR’). Herein we propose a reaction design based on a sequential process via radical and ionic paths (Figure 1C). The primary substitution of F with RO groups involves C−F bond activation by photocatalysis4g and capture of the perfluoroalkyl radical by an oxyl radical.10 Then the second transformation employs a Lewis acid and nucleophiles. Because the reaction mechanism includes an oxonium intermediate, diverse nucleophiles can be introduced. Based on our proposed design using a dual activation system, in this study we achieved a sequential C−F bond transformation of perfluoroalkylarenes via aminoxylation with a fine-tuned phenothiazine photocatalyst and aminoxyl radical reagent followed by AlCl3-mediated nucleophilic substitution with organosilicon reagents (Figure 1D).

Sequential C−F bond transformation of perfluoroalkyl compounds.
Firstly, we investigated reaction conditions for the photo-catalyzed aminoxylation using 4-phenyl(perfluorobutyl)benzene 1 a (Ered=−2.06 V vs SCE) and (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) (2) under visible light irradiation (370 nm) (Table 1). The use of Ir(ppy)3 resulted in no reaction due to the low reducing ability of excited Ir(ppy)3 (Entry 1).4g We focused on phenothiazines exhibiting higher reducing abilities than Ir(ppy)3. Their photocatalytic activities can be tuned by a substituent effect.11 N-Phenylphenothiazine PC1 exhibited a catalytic activity to mediate the aminoxylation, giving product 3 a (Entry 2). To access more negative redox potentials, phenothiazine PC2 was used, leading to an improved yield (Entry 3). It should be noted that newly developed phenothiazine PC3 bearing diisopropylamino and two methyl groups showed the best catalytic activity (Entry 4), suggesting the effect of the two Me groups is crucial to the aminoxylation. N-Dimethylphenylphenothiazine PC4 was less effective, which indicates the increase of reducing ability by an amino group is significant (Entry 5). Next, to utilize much lower reduction potential of photocatalysts, we applied thiolate catalysis12 and consecutive photo-induced electron transfer (conPET) system13 to this reaction. Thiolate catalysis resulted in no reaction, and 1 a was hardly converted (Entry 6). In the conPET system, 1 a was completely consumed, but only a trace amount of 3 a was obtained according to complicated products (Entry 7). In this case, the high reducing ability of the active catalytic species generated by conPET would cause the undesired overreduction or side-reactions. Both photocatalyst and photo-irradiation were essential for the reaction progress (Entries 8 and 9). Finally, under the optimized conditions using the 5 mol % amount of PC3, 3 a was obtained in 82 % yield (Entry 10). Further optimization for addition amount of TEMPO 2 and solvent screening is described in the Supporting Information.14
|
|||
Entry |
Catalyst |
Eox* (V vs SCE) |
Yield |
|---|---|---|---|
1 |
Ir(ppy)3 |
−1.73 |
0 % |
2 |
PC1 |
−2.45 |
35 % |
3 |
PC2 |
−2.68 |
42 % |
4 |
PC3 |
−2.57 |
60 % |
5 |
PC4 |
−2.47 |
38 % |
6[b] |
4-MeOC6H4SH |
−3.31 |
0 % |
7[c] |
Mes-Acr-BF4 |
−3.36 |
trace |
8[d] |
PC3 |
−2.57 |
0 % |
9 |
none |
– |
0 % |
10[e] |
PC3 |
−2.57 |
82 % (71 %)[f] |
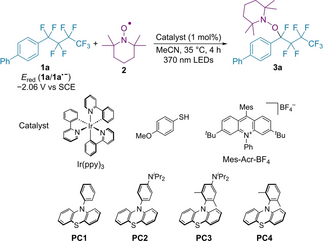
- [a] 1 a (0.4 mmol), 2 (0.8 mmol), catalyst (0.004 mmol), MeCN (2 mL), irradiation with 370 nm LEDs at 35 °C for 4 h. Yields were determined by 1H NMR spectroscopy using an internal standard. [b] 4-MeOC6H4SH (0.08 mmol), HCO2Cs (0.8 mmol), DMSO (2 mL) irradiation with 427 nm LEDs at 35 °C for 24 h. [c] Mes-Acr-BF4 (0.04 mmol), NiPr2Et (1.2 mmol), MeCN (1.3 mL), irradiation with 390 nm LEDs at 35 °C for 24 h. [d] No irradiation. [e] PC3 (0.02 mmol), 24 h. [f] Isolated yield.
Scheme 1 depicts a plausible mechanism for the aminoxylation of 1 with TEMPO 2 catalyzed by phenothiazine PC. The photoexcited PC* reduces 1 via single electron transfer (SET), affording radical anion A and radical cation PC⋅+. Mesolysis of a C−F bond affords benzyl radical B.4g Then, B associates with 2 to give product 3. PC (E(PC3⋅+/PC3)=0.61 V vs SCE) is regenerated by single electron reduction with 2 (E(2+/2)=0.62 V vs SCE).15 A by-product, N-oxoammonium cation C captures F−, suppressing the retro-reaction from B to A.16 HRMS and 19F NMR confirmed N-oxoammonium fluoride D was generated.17 The appropriate reduction potential of PC3* achieves selective reduction of starting material 1 and not product 3, realizing a single C−F bond transformation without overreduction and side-reactions.18

Proposed mechanism for photo-catalyzed aminoxylation of perfluoroalkylarenes.
We focused on the fact that our developed PC3 exhibited a more efficient catalytic activity than PC2 despite the lower reducing ability of PC3* than PC2* (Table 1).19 We conducted mechanistic studies to understand the origin of the high activity of PC3. First, the fluorescence quenching experiments of PC* with 4-trifluoromethyl(perfluorohexyl)benzene 1 e were performed. Stern–Volmer plots determined that the rate constants of the dynamic quenching of excited singlet species PC2* and PC3* were 5.3×1010 M−1 s−1 and 3.1×1010 M−1 s−1, respectively (Figures S2 and S4).20, 21 The 1st SET between 1 e and PC* is a diffusion-controlled process,22 which indicates that the catalytic turnover is independent of the reducing ability of PC*.23 We then considered the 2nd SET between PC⋅+ and TEMPO for the catalyst regeneration. The sub-microsecond transient absorption spectroscopy using laser flash photolysis method at 355 nm (4 mJ/pulse, 4 ns pulse-width) was conducted for a mixture of PC, 4-phenyl(perfluoroethyl)benzene 1 n, and TEMPO to monitor the generation and quenching of PC•+.24 The quenching rate constant of PC3⋅+ (3.2×104 M−1 s−1) was found to be much larger than that of PC2⋅+ (0.93×102 M−1 s−1) (Figures S19 and S20).25 Thus, the fast regeneration of PC3 dominates the catalytic turnover (Figure 2A). Next, Gibbs free energy changes (ΔGr) and reorganization energies (λ) in the 2nd SET were obtained by DFT calculation to estimate activation energies (ΔGǂ) according to Marcus-Hush theory (Figure 2B).26, 27 While two ΔGr values are almost identical (3.8 and 3.7 kcal/mol), λ value for PC3 (38.8 kcal/mol) is lower than that for PC2 (42.3 kcal/mol). Finally, ΔGǂ for PC3 (11.6 kcal/mol) is lower than that for PC2 (12.6 kcal/mol). This trend in ΔGǂ is consistent with that in the catalyst regeneration rate. Thus, we focused on the geometry change of PC because a smaller λ value leads to the decrease of ΔGǂ for SET. The planar structure of phenothiazine backbone in PC⋅+ changes to the bent one upon reduction (Figure 2B). This bending is the dominant geometry change and would be deeply related to λ values. We adopted the bent angle (θ) in Figure 2B right to represent the extent of a bent structure of PC. The smaller value of θ in PC3 (θ=14.5°) shows the smaller geometry change in the reduction of PC3⋅+ compared to PC2 (θ=18.0°). The steric repulsion of Me groups of PC3 suppresses bending of a phenothiazine backbone, eventually decreasing ΔGǂ. A catalyst design including both fast catalyst regeneration and effective photo-excited reduction potential is achieved by the rigidity of molecular structure and the introduction of an electron-donating group.

(A) Quenching rate constants of PC* and PC⋅+. (B) Activation energies (ΔGǂ), Gibbs free energy changes (ΔGr), reorganization energies (λ), and bent angles (θ) in the 2nd SET by DFT calculation studies ((U)ωB97XD/6-31+G(d,p)/SMD(acetonitrile)).
Using the determined optimal reaction conditions, we explored the substrate scope of this aminoxylation (Scheme 2). Electron withdrawing groups such as CN, CO2Me, and CONMe2 were available for the transformation (3 b, 3 c, 3 d). The CF3-substituted perfluoroalkylarene selectively underwent the aminoxylation in the perfluoroalkyl group (3 e).28 Silyl and boryl substituents were also tolerated (3 f and 3 g). It is noted that perfluoroalkylarenes with electron-donating groups smoothly underwent aminoxylation (3 h, 3 i, and 3 j). The development of PC3 with high reducing ability overcame the limitation of the substrate scope in our previous report for defluoroallylation.4g Substrates including pyridine, benzofuran, or naphthalene moieties efficiently gave the corresponding products (3 k, 3 l, and 3 m). The reaction of perfluoroethylarene also gave 3 n in a moderate yield. Perfluoroalkyl-substituted pyridines were feasible substrates, and various functional groups such as OMe, OH, NH2, and acetal groups were compatible with the present reaction (3 o, 3 p, 3 q, and 3 r). A quinoline-based substrate afforded desired product 3 s in 68 % yield. The substrate with two C4F9 groups underwent single aminoxylation to give product 3 t. Reactions of perfluoroalkylphenanthrene and -pyrene gave no products (3 u and 3 v).29

Substrate scope of perfluoroalkylarenes in the aminoxylation with TEMPO.[a] [a] 1 a (0.4 mmol), 2 (0.8 mmol), PC3 (0.02 mmol), MeCN (2 mL), irradiation with 370 nm LEDs at 35 °C for 24 h. Isolated yields are shown. [b] 2 a (1.2 mmol) and PC3 (0.04 mmol).
Next, Lewis acid mediators and nucleophilic coupling partners were surveyed for the selective C−F bond transformation of aminoxylated compounds 3 via an ionic path30 (Tables S3 and S4). The combination of AlCl3 and organosilicon reagents was found to be appropriate in the transformation (Scheme 3). After isolation of 3 a, which was provided by aminoxylation between 1 a and 2 (Table 1, Entry 10), 3 a was treated with allyltrimethylsilane (4 a) in the presence of AlCl3. The reaction gave allylated alcohol 5 a in 74 % yield, in which the amino group was removed on the O atom (see below). The CN and CO2Me groups were tolerated in this allylation (5 b and 5 c). Various organosilicon nucleophiles were applicable to this C−F bond transformation. Methallylsilane 4 b, silyl enol ethers 4 c and 4 d, silyl ketene acetal 4 e, and alkynylsilane 4 f provided functionalized perfluoroalkyl alcohols 5 d, 5 e, 5 f, 5 g, and 5 h, respectively. An organotin reagent, methallyltributyltin (4 g) also acted well as a nucleophile. Toluene (4 h) was a suitable nucleophile for the Friedel–Crafts reaction to give product 5 i. The reduction with HSiEt3 smoothly proceeded to yield defluorinated product 5 j. On the other hand, vinylsilane 4 j and silyl ketene imine 4 k were not applicable.

Second defluorinative transformation mediated by a Lewis acid.[a] [a] 3 (0.2 mmol), 4 (1.0 mmol), and AlCl3 (0.4 mmol) in CHCl3 (2 mL) at room temperature for 6 h. Isolated yields are shown. [b] 4 h (1 mL).
Using the radical and ionic methods to realize two types of C−F bond activation, we demonstrated a one-pot transformation of a CF2 unit via aminoxylation and allylation reactions (Scheme 4A). After aminoxylation of perfluoroalkylarene 1 a with 2 using PC3 and 370 nm LED light, the crude product was treated with 4 a and AlCl3 to give product 5 a in high yield. The one-pot aminoxylation/alkylation was also successful using silyl ketene acetal 4 e (Scheme 4B).

One-pot sequential C−F bond transformation of CF2 unit.[a] [a] 1 a (0.4 mmol), 2 (0.8 mmol), PC3 (0.02 mmol), MeCN (2 mL). Then, 4 (2.0 mmol), AlCl3 (1.2 mmol), CHCl3 (4 mL). Yields were determined by 1H NMR spectroscopy using an internal standard.
Scheme 5 illustrates a proposed mechanism for AlCl3-mediated C−F bond allylation of 3 with allylsilane 4 a. AlCl3 abstracts F− to give oxonium intermediate E. Then N-chloroamine and AlFCl2 are eliminated to give ketone F. AlCl3 activates F, and 4 a adds to a carbonyl group, affording G.31 Finally, hydrolysis of G yields product 5. The generation of F was confirmed when 3 was treated with AlCl3 in the absence of nucleophiles (Scheme S4). Other typical Lewis acids30 were not effective (Table S3). AlCl3 can mediate abstraction of fluoride ion of 3 and activation of a less basic carbonyl group of F due to high Lewis acidity. In terms of the intermediacy of ketone F, our procedure has an impact on the synthesis of perfluoroalkyl ketones from PFAS via defluorination. Traditional methods such as defluorination of fully-perfluorinated alkylbenzenes,32 perfluoroalkyl-substituted anilines33 and -enamines,34 and α-hydroperfluoroalkanoic acid esters5, 35 have problems such as narrow substrate scopes. Especially for the synthesis of aryl ketones F, available substrates were extremely limited. In this report, compounds 3 can be synthesized and used as synthetic equivalents for F with the wide substrate scope and the high compatibility of functional groups. Our process is an efficient synthetic method of functionalized perfluoroalkyl alcohols like 5 from PFAS.

Proposed mechanism for C−F bond transformation mediated by AlCl3.
In summary, a combination of photoredox catalysis and Lewis acid activation realizes sequential C−F bond transformation of a CF2 unit in perfluoroalkylarenes. Functionalized perfluoroalkyl alcohols were synthesized by phenothiazine-catalyzed photo-induced defluoroaminoxylation with TEMPO and subsequent AlCl3 mediated substitution of a F atom with various carbon nucleophiles. Mechanistic studies revealed that the rigidity of molecular structure and the introduction of an electron-donating group is important in catalyst design to achieve fast catalyst regeneration and effective photo-excited reduction potential.
Acknowledgments
This work was financially supported by JSPS KAKENHI (JP23K17845 (M.Y.), JP23H04906 (M.F.)). This work was supported by the Grant-in-Aid for Transformative Research Areas (A) JP21H05212 (M.Y.) Digitalization-driven Transformative Organic Synthesis (Digi-TOS) from the Ministry of Education, Culture, Sports, Science & Technology, Japan and JST CREST Grant No. JPMJCR20R3 (M.Y.) and JPMJCR18R4 (M.A.), Japan. We thank the members of the Radiation Laboratory of SANKEN, Osaka University, for running the linear accelerator. N.S. acknowledges support from the JSPS Fellowship for Young Scientists (JP22J10775). Y.N. acknowledges support from Research Encouragement Grants of The Asahi Glass Foundation.
Conflict of interests
The authors declare no conflict of interest.
Open Research
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.

