

Tris(ortho-carboranyl)borane: An Isolable, Halogen-Free, Lewis Superacid
Abstract
The synthesis of tris(ortho-carboranyl)borane (BoCb3), a single site neutral Lewis superacid, in one pot from commercially available materials is achieved. The high fluoride ion affinity (FIA) confirms its classification as a Lewis superacid and the Gutmann-Beckett method as well as adducts with Lewis bases indicate stronger Lewis acidity over the widely used fluorinated aryl boranes. The electron withdrawing effect of ortho-carborane and lack of pi-delocalization of the LUMO rationalize the unusually high Lewis acidity. Catalytic studies indicate that BoCb3 is a superior catalyst for promoting C−F bond functionalization reactions than tris(pentafluorophenyl)borane [B(C6F5)3].
Boranes are useful Lewis acids in stoichiometric and catalytic reactions by taking advantage of the vacant p-orbital.1 Boron trihalides (BX3; X=F, Cl, Br) are ubiquitous examples; however, their volatile nature and labile B−X bonds make them incompatible with many substrates that limits their utility. In this regard, tris(pentafluorophenyl)borane [B(C6F5)3, Figure 1], became a standout borane due to excellent thermal stability and functional group tolerance which led to its widespread use as a Lewis acid reagent and catalyst.2 Although B(C6F5)3 is a useful Lewis acid, its’ fluoride ion affinity (FIA) is less than the defined benchmark Lewis superacid (LSA), SbF5.3, 4 In fact, reports on single-site LSA boranes are scarce and accessing new species with different acidity or steric profiles could lead to altered reactivity and selectivity in stoichiometric or catalytic reactions. Although there have been efforts directed at isolating boron based Lewis superacids, most of the methods involve modifications on the substitution of the aryl group on triarylboranes.2b This strategy was effective in achieving LSA for B(p-CF3-C6F4)3 while other fluoride loading approaches fell short of the LSA criterion.5 The increased FIA of B(p-CF3-C6F4)3 results in greater catalytic activity in C−H perfluoroalkylation and perfluoroarylation reactions compared to B(C6F5)3.6 Incorporating the boron center within an anti-aromatic heterocycle results in compounds with high Lewis acidity based on Gutmann-Beckett acceptor numbers, or high FIAs, but their reactive B−C bonds limits application in synthesis.,[7][8] Fluorinated alkyloxy-boranes, boron pseudohalides [B(ORF)3, RF=−CF3, −TeF5, −SO2CF3], and perfluoroalkyl-boranes are known and believed to be LSAs; however, they are thermally unstable with the latter only generated as fleeting species.9, 10 Chlorination approaches in triarylboranes have been extensively studied with little success.11 Gabbaï and co-workers applied a different approach, appending cationic substituents (trialkylammonium, sulfonium, and phosphonium) on aryl boranes as an alternative electron withdrawing group to generate a class of cationic Lewis acids with high FIAs.12 Recently, Berionni and co-workers achieved remarkable FIAs with rigid pyramidalized boranes tethered by carbon, phosphonium and sulfonium centers, although the free boranes were not isolable.13
We postulated that using an unconventional electron withdrawing group with significant steric protection could result in the isolation of a new class of trigonal planar Lewis acids. In the literature, carboranes14 offer both of these attributes although they have been minimally explored in this realm.15, 16 The C2B10 carborane cluster is exceptionally stable and the three-dimensional icosahedron presents a sphere-like steric profile in stark contrast to flat aryl groups. Within the C2B10 carboranes, three isomers exist with each classified based on the relative positioning of the carbon atoms, ortho- (adjacent), meta- (one boron betwixt), and para- (carbon atoms on opposite sides of the icosahedron). Among these, the ortho-isomer (oCbH) has been documented as the most electron withdrawing if C-bound. This hypothesis is supported by a report of borole analogues featuring a bis(ortho-carborane) backbone in which the o-carborane engendered significantly higher Lewis acidity than the biphenyl variant.17 A notable advancement in this realm was the preparation of anthracene analogues by Ye and co-workers in which the boron centers are bridging two ortho-carboranes.18 In these species, LSA is achieved but the halides or azides present on the boron center are reactive and these fall into the category of dual site LSAs.19 In contrast to aryl or fluoroaryl boranes, the carborane cluster is likely unable to delocalize the LUMO, primarily a p-orbital on boron. We herein report the synthesis of BoCb3 in one pot which represents an isolable, halogen-free, neutral, single-site Lewis superacid; and investigate its properties.

Tris(pentafluorophenyl)borane, isolable single-site neutral boron Lewis superacids (LSAs) in the literature and feature complex, tris(ortho-carboranyl)borane (oCb=ortho-carborane, C2B10H11).
The lithiation of ortho-carborane with nBuLi and subsequent reaction with 0.33 equivalents of BCl3 (1 M solution in hexanes) generated BoCb3 in 29 % yield after a work up (Scheme 1). When BBr3 was used instead of BCl3, an increased isolated yield of 35 % was achieved. The structure was confirmed by a single-crystal X-ray diffraction study (Figure 2).20 The geometry is trigonal planar as all C−B−C bond angles are within error of 120° and the C−B bond lengths are slightly longer than the typical C(o-caroborane)−B single bond in boranes species [1.614(8)–1.627(7) Å c.f. ≈1.58 Å], which is attributed to the bulk around the central B-atom. The downfield 11B{1H} resonance at 67.2 ppm is assigned to the central tricoordinate boron atom and peaks ranging from 7.4 to −12.4 ppm to the cluster atoms. The C−H protons are the most diagnostic in the 1H NMR spectrum and appear as a singlet at 5.02 ppm while the corresponding carbon is observed in the 13C{1H} NMR spectrum at 65.0 ppm and a broad peak at 69.3 ppm is assigned to the carbon bound to the central boron. Analytical purity was confirmed by microanalysis and the melting point exceeds 250 °C, indicating thermal stability.

Synthesis of BoCb3. [Isolation: After the reaction was complete, additional toluene was added and the reaction mixture filtered through a small pad of celite and washed with dichloromethane. The volatiles were stripped from the combined filtrate under vacuum and diethyl ether was added to make a suspension, which was filtered to give pure BoCb3 as white solid.]

Solid state structure of BoCb3. Ellipsoids depicted at the 50 % probability level and hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): B(1)−C(1) 1.614(8), B(1)−C(3) 1.627(7), B(1)−C(5) 1.626(8); C(1)−B(1)−C(3) 119.8(5), C(1)−B(1)−C(5) 120.3(5), C(3)−B(1)−C(5) 119.9 (5).
To experimentally investigate the relative Lewis acidity, the corresponding acetonitrile [CH3CN ⋅ BoCb3] and benzaldehyde [PhCHO ⋅ BoCb3] adducts were prepared (Scheme 2). In the 11B NMR spectra, the peak for the central boron atom (67.2 ppm) shifts to the tetracoordinate region among the cluster boron peaks. The C−H resonance of BoCb3 (5.02 ppm) in the 1H NMR spectrum shifts upfield to 4.60, and 4.77 for CH3CN ⋅ BoCb3 and PhCHO ⋅ BoCb3, respectively. Their identities were further confirmed by single crystal X-ray diffraction experiments (Figure 3) with the central boron atoms adopting distorted tetrahedral geometries. The shorter B-donor atom (O, and N) bonds in the BoCb3-adducts compared to the corresponding B(C6F5)3-complexes indicate higher Lewis acidity of BoCb3 [CH3CN ⋅ BoCb3: B−N 1.5679(17) Å c.f. CH3CN ⋅ B(C6F5)3: B−N 1.616(3) Å; PhCHO ⋅ BoCb3: B−O 1.551(2) Å, c.f. PhCHO ⋅ B(C6F5)3: B−O 1.6108(8) Å].21 Although the structure of the PhCHO ⋅ BoCb3 was confirmed in the solid state, we observed a dissociation equilibrium in solution which could not be resolved in the 1H NMR spectrum at 25 °C. The 1H NMR spectra of BoCb3 with gradient PhCHO concentrations at 25 °C and variable temperature studies of 1 : 1 and 1 : 1.4 molar mixtures of BoCb3 and PhCHO confirm the dissociation equilibria (see Supporting Information Figures S21–S23). The CN stretching frequency is often used as a parameter to evaluate Lewis acidity. However, the CN stretching frequencies of CH3CN ⋅ BoCb3 (2363 cm−1) and CH3CN ⋅ B(C6F5)3 (2367 cm−1) are within experimental error. To find the relative Lewis acidity between BoCb3 versus B(C6F5)3, a competition study was performed to assess the relative binding affinity of acetonitrile. When equimolar BoCb3, B(C6F5)3, and CH3CN were mixed in CDCl3, CH3CN ⋅ BoCb3 and CH3CN ⋅ B(C6F5)3 were observed in a ≈2 : 1 ratio indicating higher CH3CN affinity of BoCb3 over B(C6F5)3.22

Synthesis of nitrile- [CH3CN ⋅ BoCb3] and aldehyde- [PhCHO ⋅ BoCb3] adducts of BoCb3.

Solid state structure of adducts CH3CN ⋅ BoCb3, PhCHO ⋅ BoCb3, and Et3PO ⋅ BoCb3 (left to right). Ellipsoids depicted at the 50 % probability level, hydrogen atoms, and solvates are omitted for clarity. Selected bond lengths (Å) and angles (°) of CH3CN ⋅ BoCb3: B(1)−N(1) 1.5679(17), N(1)−C(7) 1.1380(18), B(1)−C(1) 1.714(2), B(1)−C(3) 1.7118(19), B(1)−C(5) 1.7143(19), B(1)−N(1)−C(7) 176.44(13); PhCHO ⋅ BoCb3: B(1)−O(1) 1.551(2), O(1)−C(7) 1.254(3), B(1)−C(1) 1.714(3), B(1)−C(3) 1.718(3), B(1)−C(5) 1.706(3); B(1)−O(1)−C(7) 134.57(18), O(1)−C(7)−C(8) 123.2(2); Et3PO ⋅ BoCb3: B(1)−O(1) 1.500(2), O(1)−P(1) 1.5344(11), B(1)−C(1) 1.730(2), B(1)−C(3) 1.732(2), B(1)−C(5) 1.730(2); B(1)−O(1)−P(1) 177.85(11).
The Gutmann-Beckett method was applied to evaluate the Lewis acidity of BoCb3 by synthesizing Et3PO ⋅ BoCb3 (Table 1).23 The difference in 31P chemical shift (Δδ1) of Et3PO ⋅ BoCb3 and Et3PO is 34.1 ppm, 27.5 ppm, and 30.1 ppm in C6D6, CDCl3, and CD2Cl2, respectively (Table 1).24 These differences in chemical shifts are higher than the results obtained for B(C6F5)3 (Δδ2=29.7 ppm in C6D6, 23.5 ppm in CDCl3 and 26.1 ppm in CD2Cl2) and those reported for B(p-CF3-C6F4)3 (Δδ2=31.9 ppm in C6D6 and Δδ2=29.0 ppm in CD2Cl2).5 This indicates that BoCb3 is more Lewis acidic than B(C6F5)3 and B(p-CF3-C6F4)3 based on the Gutmann-Beckett method. Also, the shorter B−O bond in Et3PO ⋅ BoCb3 [B−O 1.500(2) Å] over Et3PO ⋅ B(C6F5)3 [B−O 1.533(3) Å] imply a stronger bond in the BoCb3 adduct (Figure 3).25
|
|||||
Solv. |
Et3PO [δ 31P] |
Et3PO ⋅ BoCb3 [δ 31P] |
Et3PO ⋅ B(C6F5)3 [δ 31P] |
Et3PO ⋅ BoCb3-Et3PO [Δδ1] |
Et3PO ⋅ B(C6F5)3-Et3PO [Δδ2] |
|---|---|---|---|---|---|
C6D6 |
45.7 |
79.6 |
75.4 |
34.1 |
29.7 |
CDCl3 |
52.3 |
79.8 |
75.8 |
27.5 |
23.5 |
CD2Cl2 |
51.0 |
81.1 |
77.1 |
30.1 |
26.1 |

To support the experimental observations on the high Lewis acidity of BoCb3 we undertook a variety of theoretical analyses against benchmark strong Lewis acidic boranes, specifically B(p-CF3-C6F4)3 and B(C6F5)3 (Table 2). Considered parameters include fluoride ion affinity (FIA), hydride ion affinity (HIA), binding energy to the Lewis bases NH3 and CH3CN, Natural Population Analysis Charges, relative LUMO energies, and finally, the Global Electrophilicity Index (GEI) recently reported by Stephan and co-workers.26
Compound |
FIA (Krossing) [ΔH, kJ mol−1] |
HIA (Krossing) [ΔH, kJ mol−1] |
NH3 affinity [ΔG, kJ mol−1] |
CH3CN affinity [ΔG, kJ mol−1] |
LUMO [eV] |
GEI |
|---|---|---|---|---|---|---|
BoCb3 |
605 |
622 |
149 |
89 |
−3.99 |
4.22 |
B(C6F5)3 |
452 |
484 |
97 |
33 |
−3.50 |
3.78 |
B(p-CF3-C6F4)3 |
501 |
537 |
110 |
51 |
−4.09 |
4.79 |
SbF5 |
493 |
– |
– |
– |
– |
– |
BF3 |
– |
299 |
– |
– |
– |
– |
Calculations for gas phase fluoride and hydride affinities are beset with issues in obtaining values consistent with experiment due to the small size and hardness of the anions and also the lack of a consistent referencing system. Krossing and co-workers presented an isodesmic reaction benchmarked to accurate fluoride and hydride affinities for [(CH3)3Si]+ combined with inexpensive calculations using BP86/SVP for the single point calculations for Lewis acids.27 Gas phase values for direct addition of F− and H− using B3LYP−D3/def2SVP are presented in the Supporting Information. Antimony pentafluoride has been established as the threshold LSA, with a calculated fluoride ion affinity (FIA) using Krossing's method of 493 kJ mol−1.27 Using Krossing's method benchmarked to their result for B(C6F5)3 at 452 kJ mol−1, we verified our calculations to reproduce their result for SbF5 at 493 kJ mol−1. The calculated FIA of BoCb3 is 605 kJ mol−1 and B(p-CF3-C6F4)3 is 501 kJ/mol, indicating that BoCb3 has a significantly higher FIA than the other two boranes and SbF5, classifying it as a LSA. To validate the theoretically determined high FIA, BoCb3 was reacted with [nBu4N][SbF6] aiming to abstract a fluoride ion from SbF6−. The 1 : 1 reaction of BoCb3 and [nBu4N][SbF6] at room temperature in CDCl3 resulted in consumption of BoCb3 and SbF6− within 30 min indicating fluoride ion abstraction based on 1H, 19F{1H}, and 11B{1H} NMR spectroscopy.22
For hydride ion affinities, using Krossing's method, anchoring to their results for B(C6F5)3 (543 kJ mol−1) and BF3 (299 kJ mol−1), again BoCb3 has the largest HIA of the considered Lewis acids at 622 kJ mol−1. Calculating affinities for neutral Lewis bases (B3LYP-D3/def2SVP, gas phase), BoCb3 also has a larger affinity with a ΔG for the dissociation of NH3 being 149 kJ mol−1, compared to 110 and 97 kJ mol−1 for B(p-CF3-C6F4)3 and B(C6F5)3, respectively. For acetonitrile, the ΔG for dissociation was calculated at 89 kJ mol−1 for BoCb3, 33 kJ mol−1 for B(C6F5)3, and 51 kJ mol−1 for B(p-CF3-C6F4)3, qualitatively consistent with the experimental observation that BoCb3 outcompetes B(C6F5)3 for acetonitrile. For all affinity calculations, the results indicate BoCb3 is the strongest Lewis acid, followed by B(p-CF3-C6F4)3, then B(C6F5)3. However, the LUMO of B(p-CF3-C6F4)3, calculated using B3LYP-def2TZVP to give results directly comparable to the literature,26 is 0.1 eV lower in energy than BoCb3, which also has a correspondingly larger Global Electrophilicity Index at 4.79 compared to 4.22 for BoCb3. The discrepancy between the orbital energy difference in B(p-CF3-C6F4)3 and BoCb3 versus the thermochemical Lewis acidity predictions are rationalized by a qualitative examination of the LUMOs themselves, depicted in Figure 4. The LUMO for BoCb3 is entirely localized on the boron atom, whereas the LUMO for B(p-CF3-C6F4)3 [and also B(C6F5)3] is partially delocalized into the arene rings. Increased localization of the LUMO of BoCb3 is also reflected in the Natural Population Charges on the boron centers, where the central boron BoCb3 carries a positive charge of +1.83. The central boron atoms in B(p-CF3-C6F4)3 and B(C6F5)3 carry positive charges of +0.81 and +0.78, respectively, that is likely due to the phenyl rings ability to donate some electron density to the central atom.

Depictions of the LUMO for BoCb3, B(C6F5)3, and B(p-CF3-C6F4)3 [from left to right, B3LYP/def2TZVP calculations]
The steric profiles and buried volume with respect to the boron centers for BoCb3 and B(C6F5)3 were compared using the SambVca 2.1 routine28 on the calculated optimized structures. The z-axis was defined as perpendicular to the BR3 trigonal plane. It was found that the carborane substituents result in greater steric demand at B with a buried volume of 92.2 % compared to a buried volume of 83.5 % for B(C6F5)3.22
Next, we evaluated BoCb3 as a Lewis acid in catalytic transformations. The abnormally high fluoride ion affinity encouraged us to explore it as a catalyst in C−F bond activation reactions.29 The catalytic functionalization of C−F bonds is challenging given that C−F is the strongest carbon single bond. To check the efficacy of BoCb3, we attempted a model hydrodefluorination30 reaction of 1-fluoroadamantane (1-F−Ad) and triethylsilane. To our delight, when one equivalent of 1-F−Ad was treated with 1.1 equivalents of HSiEt3 in the presence of 0.5 mol% BoCb3 in CDCl3 at 23 °C for 10 minutes, the reduction product, adamantane (Ad) was obtained in quantitative yield (>97 % yield by NMR spectroscopy) along with FSiEt3 as the side product (Table 3, entry 1). Interestingly, when 0.5 mol% B(C6F5)3 was employed (entry 2), adamantane (Ad), was only generated in 60 % yield based on NMR spectroscopy, along with unreacted starting materials (Table 3, entry 1). Longer reaction times did not change the outcome. However, Stephan and co-workers showed that B(C6F5)3 can undergo the same transformation but in a tenfold increase in catalyst loading (5 mol%).31 It is known that B(C6F5)3 reacts with HSiEt3 upon heating to give HB(C6F5)2 (Piers’ borane)32 but surprisingly, BoCb3 is unreactive with HSiEt3 ruling out the possibility of HBoCb2 being the active catalyst.33 When BoCb3 was reacted with 1-F-Ad, both starting materials were consumed resulting in a complex mixture based on 1H, 19F{1H}, and 11B{1H} NMR spectroscopy. This suggests that fluoride abstraction is the first step in the mechanism of the catalytic transformation.22
|
|||
Entry |
“Conditions” |
Product (1-Nu-Ad) |
Yield |
|---|---|---|---|
1 |
1.1 equiv HSiEt3, 0.5 mol% BoCb3, CDCl3 (1 mL), 23 °C, 10 min |
Ad |
>97 %[a] |
2 |
1.1 equiv HSiEt3, 0.5 mol% B(C6F5)3, CDCl3 (1 mL), 23 °C, 30 min |
Ad |
60 %[a] |
3 |
1 mol% BoCb3, C6H6 (2 mL), 23 °C, 30 min |
1-Ph-Ad |
90 %[b] |
4 |
1 mol% B(C6F5)3, C6H6 (2 mL), 23 °C, 4 h |
1-Ph-Ad |
NR[c] |
5 |
1 mol% BoCb3, C7H8 (2 mL), 23 °C, 30 min |
1-Tol-Ad |
72 %[b] |
6 |
1 mol% BoCb3, m-Xylene (2 mL), 23 °C, 30 min |
1-Xyl-Ad |
92 % [b] |
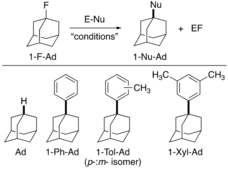
- [a] Yields are determined by NMR spectroscopy using 1-bromo-4-fluorobenzene as an internal standard. [b] Isolated yields. [c] NR=No reaction.
Friedel–Crafts reactions of 1-F-Ad were also investigated. When 1-F-Ad was reacted with benzene in the presence of 1 mol% BoCb3, 1-Ph-Ad was obtained in 90 % isolated yield after 30 min (Table 3, entry 3). Interestingly, when B(C6F5)3 was employed in the same transformation, we did not observe any product formation even after 4 h (entry 3 and 4). Moran and co-workers reported a H2O ⋅ B(C6F5)3-catalyzed Friedel–Crafts reaction of 1-F-Ad but highly electron rich arenes and specific solvent combinations were required to enable the reaction.34 The analogous transformation with toluene and m-xylene provides the corresponding products 1-Tol-Ad (entry 5) in 72 % isolated yield (20 : 3 p-:m- isomeric ratio), and 1-Xyl-Ad selectively (entry 6) in 92 % yield. The improved outcome in the reactions of BoCb3 over B(C6F5)3 is attributed to its higher FIA and Lewis acidity. The catalytic hydrosilylation reaction2c of benzaldehyde with triethylsilane (1 : 1 PhCHO and HSiEt3 in presence of 5 mol% BoCb3 in CDCl3) formed a mixture of the hydrosilylation product PhCH2OSiEt3 and dialkyl ether (PhCH2)2O in 52 % and 36 % yield by NMR spectroscopy at room temperature after 24 h.22
In summary, the halogen-free thermally stable borane, tris(ortho-carboranyl)borane, is accessed from three convenient reagents: ortho-carborane, nBuLi, and BBr3. Calculated fluoride ion affinities reveal the Lewis acidity greatly exceeds SbF5 making it a rare example of a Lewis superacidic borane. Theoretical binding studies comparing BoCb3 to the state-of-the-art fluoroarylborane Lewis acids, B(C6F5)3 and B(p-CF3-C6F4)3, reveal fluoride and hydride ion affinities as well as Lewis base affinities of ammonia and acetonitrile are all the highest for BoCb3. Experimentally, a competition study in solution with acetonitrile and one equivalent of BoCb3 and B(C6F5)3 corroborate preference for binding BoCb3. In the solid state, X-ray diffraction data from benzaldehyde, acetonitrile and triethylphosphine oxide adducts all validate stronger binding to BoCb3 than B(C6F5)3. In solution, Gutmann-Beckett studies indicate the Lewis acidity of BoCb3 exceeds the literature values for B(C6F5)3 and B(p-CF3-C6F4)3. The high FIA can be taken advantage of in catalytic C−F bond activation reactions of unactivated alkyl fluorides by reduction with triethylsilane and C−C bond forming Friedel–Crafts reactions with arenes. In this disclosure of BoCb3, we merely touch on the potential of this reagent and catalyst. Given the powerful Lewis acidity and unusual steric profile, it offers unique synthetic opportunities in boron mediated transformations.
Acknowledgements
We are grateful to Baylor University, the Welch Foundation (Grant No. AA-1846), the National Science Foundation (Award No. 1753025), and the Australian Research Council (FT16010007, DP20010013) for their support of this work. Allocation of computing resources from La Trobe University are acknowledged. We thank Tyler Bartholome for preliminary studies. We appreciate the gift of ortho-carborane used in this study from the late F. Gordon A. Stone [inventor of B(C6F5)3], Bruce Hodson, and Thomas McGrath. Xianzhong Xu is thanked for assistance with NMR spectroscopic experiments.
Conflict of interest
The authors declare no conflict of interest.
Open Research
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.

